研究室紹介Laboratories
- Back
- Top > 研究室紹介 > がん分子病因・病態学(連携) > がんシステム情報学(愛知県がんセンター)
がん分子病因・病態学(連携)がんシステム情報学(愛知県がんセンター)
概要
![]()
本講座は、がんの複雑なシステムをゲノム情報等の大規模パーソナルオミクスプロファイルデータの解析を通じて理解し、得られた知見を、個人に最適な予防や治療等に役立てることを目指して研究を行っています。そのために、深層学習モデリングやベイズ統計モデリング等の、先進的なデータ科学技術に基づくデータ解析手法を開発し、スーパーコンピュータを用いた解析にフル活用しています。また基礎的なデータ解析手法の開発に加え、がんゲノム医療の現場で喫緊の課題となっている、エキスパートパネルにおける医療者の判断の精緻化・省力化に資する人工知能を活用した情報解析基盤技術の開発を進めています。
研究プロジェクト
1) ベイズ統計モデリングに基づく高精度体細胞変異検出技術の開発
DNAシークエンスリードデータからの、体細胞変異検出(変異コール)において、リード中に含まれる読み取りエラーと真の変異情報を分離し、高精度に変異を検出するための、ベイズ統計モデリング技術に基づく手法を開発しています。このような高精度変異検出技術は、全ゲノムシークエンスデータのように、シークエンス読み取り深度の浅いデータからの変異検出や、低頻度変異アレル情報により規定される、がん細胞クローン集団構造の決定ために重要です。
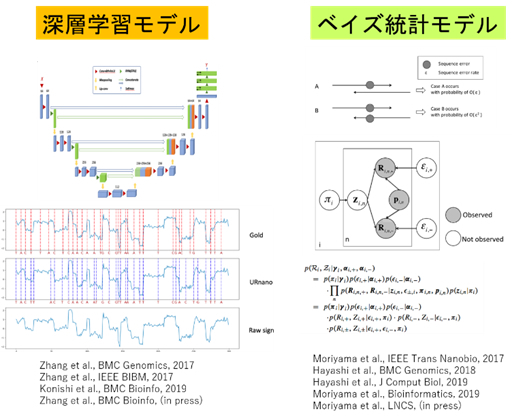
上記の目的のために、従来の変異検出プロセスにおいて見逃されてきた、リード中に含まれる補助的な情報を活用する手法を研究してきました。例えば、あるペアードシークエンスリードの組が、同じDNAフラグメントを重複して計測した領域における変異候補パターンの情報を用いて、エラーと変異を高精度に分離する階層型ベイズモデル(OVarCall; Moriyama et al., 2017)を開発しました。また、他の種類の補助的情報を活用した変異検出モデルが複数存在する場合に、それらのモデル群からの情報を合理的に統合するベイズ統計的枠組み(OHVarfinDer; Moriyama et al., 2019a)を開発し、更なる変異検出精度の向上を示しました。また同一個人の複数領域シークエンスデータ中の変異候補情報を統合する、ベイズ型変異検出モデル(MultiMuc; Moriyama et al., 2019b)を開発し、各検体における変異検出の精度向上に成功しています。
2) 免疫細胞シークエンスデータ解析技術の開発
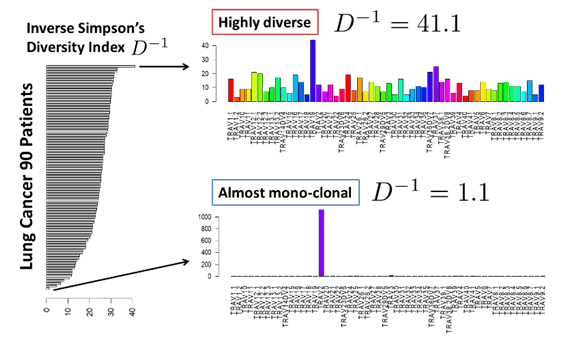
がんと免疫の関係を解き明かし、治療に役だてることは近年のがん医療における大きな課題です。そのために、がんと免疫に関わる様々なモダリティを持つデータから有用情報を抽出するための手法群を開発しています。
一例として、T細胞受容体(TCR)やB細胞受容体(BCR)配列で特徴づけられる、免疫細胞クローンレパトア解析のためのシークエンスデータ解析パイプライン(TCRip/BCRip; Fang, Yamaguchi, et al., 2014他)を開発しています。ここではTCRまたはBCRのRNAシークエンスデータから、リードの部分配列アライメントを元に、高精度に各クローンを特徴づける配列を決定し、レパトア構造を推定するアルゴリズムを考案しています。同パイプラインは、がんワクチンや免疫チェックポイント阻害剤治療前後のレパトア構造の動的変化の解析等に活用されています。
また他に、DNAシークエンスデータからのHLA遺伝子型を高精度に決定する、階層型ベイズモデル(ALPHLARD; Hayashi et al., 2018)の開発を行っています。本手法は、読み取り深度が浅い全ゲノムシークエンスデータにおいても、既存の手法を大幅に上回る精度で、HLAの型決定を行うことが出来ます。また他の多くの手法において、型決定を行うことができないHLA クラスII遺伝子の型決定も行うことができます。同手法を用いて、決定されたHLA型情報は、ICGC Pacncancer Analysis of Whole Genomes (PCAWG) Project内の共通リソースとして活用されています。また、同手法を改良し、がん細胞、正常細胞のペアのシークエンスデータの情報を同時に考慮するモデル(ALPHLARD-NT; Hayashi et al., 2019)を開発しています、その結果、更なるHLA遺伝子型決定の精度の向上を得るとともに、HLA遺伝子中の体細胞変異の高精度検出にも成功しています。
3) 深層学習技術に基づくシークエンスデータ解析技術の開発
人間の先験的知識によるモデル化や特徴抽出が困難な問題において、深層学習モデルは有効です。我々はシークエンスデータからの情報抽出において、深層ニューラルネットワークモデルを活用した手法を開発研究してきています。例えば、RNA-seqデータに含まれる様々なバイアスの補正にリカレントニューラルネットワークを用いた手法を考案しています(Zhang et al., 2017a)。コピー数変異などの大きな構造変異を高速かつ高精度に決定するためのモデルも研究している(Zhang et al., 2017b)。
またナノポアシークエンサーからの、ロングリードが活用され始めていますが、既存のショートリードシークエンサーからのデータに比べて正確性が低いことが問題となっています。これは、ナノポアをDNAストランドが通過するときに検出される電流の値を、正確にDNAの塩基配列に変換できないからです。我々は、この問題に対して、新たな深層ニューラルネットワークモデル(URnano; Zhang et al., (in press))を提案し、既存の手法によりも高い精度で塩基配列への変換を果たしています。
4)がんゲノム臨床シークエンスのための情報解析基盤技術の開発
がんゲノムパネル検査が、2019年より保険適用となり、がんゲノム医療が本格化しようとしています。我々は、近未来の、全ゲノムシークエンスおよび複数オミックスデータの統合解析に基づく、がん臨床シークエンスの実現に向けた情報解析基盤技術の開発を進めています。
臨床シークエンスにおいて喫緊の課題は、高速のデータ解析に加え、ゲノム解析の結果得られた変異情報を臨床上有用な情報へ、迅速かつ正確に解釈・翻訳し、エキスパートパネルにおける意思決定の精緻化につなげることです。そのために人工知能を活用した解釈システムの研究を進めている。当分野は、愛知県がんセンター病院のエキスパートパネルへも参画しており。今後、現場からのフィードバックを活用して、より実践的かつ未来を見据えたシステムの開発を進めていく予定です。
教員
| 構成員名 | 役職 | 所属 |
|---|---|---|
| 山口 類 | 連携教授 | 愛知県がんセンター |
研究実績
- 2019年
- Zhang YZ, Akdemir A, Tremmel G, Imoto S, Miyano S, Shibuya T, Yamaguchi R. Nanopore base-calling from a perspective of instance segmentation. BMC Bioinformatics. (in press)
- Ito S, Yadome M, Nishiki T, Ishiduki S, Inoue H, Yamaguchi R, Miyano S. Virtual Grid Engine: A simulated grid engine environment for large-scale supercomputers. BMC Bioinformatics. (in press)
- Kasajima R, Yamaguchi R, Shimizu E, Tamada Y, Niida A, Tremmel G, Kishida T, Aoki I, Imoto S, Miyano S, Uemura H, Miyagi Y. Variant analysis of prostate cancer in the Japanese and a new attempt to predict related biological pathways. Oncology Reports. (in press)
- Yoshino T, Katayama K, Yamaguchi R, et al. Classification of patients with cold sensation by a review of systems database: A single-centre observational study. Complement Ther Med. 2019;45:7-13.
- Yamaguchi K, Shimizu E, Yamaguchi R, et al. Development of an MSI-positive colon tumor with aberrant DNA methylation in a PPAP patient. J Hum Genet. 2019;64(8):729-740.
- VanderWeele DJ, Finney R, Katayama K, et al. Genomic Heterogeneity Within Individual Prostate Cancer Foci Impacts Predictive Biomarkers of Targeted Therapy. Eur Urol Focus. 2019;5(3):416-424.
- Tsuda Y, Hirata M, Katayama K, et al. Massively parallel sequencing of tenosynovial giant cell tumors reveals novel CSF1 fusion transcripts and novel somatic CBL mutations. Int J Cancer. 2019;145(12):3276-3284.
- Takeda R, Yokoyama K, Ogawa M, et al. The first case of elderly TCF3-HLF-positive B-cell acute lymphoblastic leukemia. Leuk Lymphoma. 2019;60(11):2821-2824.
- Takeda R, Yokoyama K, Kobayashi S, et al. An Unusually Short Latent Period of Therapy-Related Myeloid Neoplasm Harboring a Rare MLL-EP300 Rearrangement: Case Report and Literature Review. Case Rep Hematol. 2019;2019:4532434.
- Nakamura S, Yokoyama K, Shimizu E, et al. Prognostic impact of circulating tumor DNA status post-allogeneic hematopoietic stem cell transplantation in AML and MDS. Blood. 2019;133(25):2682-2695.
- Muraoka D, Seo N, Hayashi T, et al. Antigen delivery targeted to tumor-associated macrophages overcomes tumor immune resistance. J Clin Invest. 2019;129(3):1278-1294.
- Moriyama T, Imoto S, Hayashi S, Shiraishi Y, Miyano S, Yamaguchi R. A Bayesian model integration for mutation calling through data partitioning. Bioinformatics. 2019;35(21):4247-4254.
- Maeda-Minami A, Yoshino T, Katayama K, et al. Prediction of deficiency-excess pattern in Japanese Kampo medicine: Multi-centre data collection. Complement Ther Med. 2019;45:228-233.
- Konishi H, Komura D, Katoh H, et al. Capturing the differences between humoral immunity in the normal and tumor environments from repertoire-seq of B-cell receptors using supervised machine learning. BMC Bioinformatics. 2019;20(1):267.
- Hosono Y, Masuishi T, Mitani S, et al. Evaluation of ALK Fusion Newly Identified in Colon Cancer by a Comprehensive Genomic Analysis. JCO Precision Oncology. 2019(3):1-5.
- Hayashi S, Moriyama T, Yamaguchi R, et al. ALPHLARD-NT: Bayesian Method for Human Leukocyte Antigen Genotyping and Mutation Calling through Simultaneous Analysis of Normal and Tumor Whole-Genome Sequence Data. J Comput Biol. 2019;26(9):923-937.
- Hasegawa T, Yamaguchi R, Niida A, Miyano S, Imoto S. Ensemble Smoothers for Inference of Hidden States and Parameters in Combinatorial Regulatory Model. Journal of the Franklin Institute. 2019.
- 2018年
- Yokoyama K, Shimizu E, Yokoyama N, et al. Cell-lineage level-targeted sequencing to identify acute myeloid leukemia with myelodysplasia-related changes. Blood Adv. 2018;2(19):2513-2521.
- Takei T, Yokoyama K, Shimizu E, et al. Azacitidine effectively reduces TP53-mutant leukemic cell burden in secondary acute myeloid leukemia after cord blood transplantation. Leuk Lymphoma. 2018;59(11):2755-2756.
- Ogawa M, Yokoyama K, Hirano M, et al. Different clonal dynamics of chronic myeloid leukaemia between bone marrow and the central nervous system. Br J Haematol. 2018;183(5):842-845.
- Nakamura S, Yokoyama K, Yusa N, et al. Circulating tumor DNA dynamically predicts response and/or relapse in patients with hematological malignancies. Int J Hematol. 2018;108(4):402-410.
- Kobayashi M, Yokoyama K, Shimizu E, et al. Phenotype-based gene analysis allowed successful diagnosis of X-linked neutropenia associated with a novel WASp mutation. Ann Hematol. 2018;97(2):367-369.
- Kiyotani K, Mai TH, Yamaguchi R, et al. Characterization of the B-cell receptor repertoires in peanut allergic subjects undergoing oral immunotherapy. J Hum Genet. 2018;63(2):239-248.
- Ito S, Yadome M, Nishiki T, et al. Virtual Grid Engine: Accelerating thousands of omics sample analyses using large-scale supercomputers. Paper presented at: 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM); 3-6 Dec. 2018, 2018.
- Inoue D, Fujino T, Sheridan P, et al. A novel ASXL1-OGT axis plays roles in H3K4 methylation and tumor suppression in myeloid malignancies. Leukemia. 2018;32(6):1327-1337.
- Hayashi S, Yamaguchi R, Mizuno S, et al. ALPHLARD: a Bayesian method for analyzing HLA genes from whole genome sequence data. BMC Genomics. 2018;19(1):790.
- 2017年
- Zhang YZ, Yamaguchi R, Imoto S, Miyano S. Sequence-specific bias correction for RNA-seq data using recurrent neural networks. BMC Genomics. 2017;18(Suppl 1):1044.
- Zhang YZ, Imoto S, Miyano S, Yamaguchi R. Reconstruction of high read-depth signals from low-depth whole genome sequencing data using deep learning. Paper presented at: 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM); 13-16 Nov. 2017, 2017.
- Tsuda Y, Tanikawa C, Miyamoto T, et al. Identification of a p53 target, CD137L, that mediates growth suppression and immune response of osteosarcoma cells. Sci Rep. 2017;7(1):10739.
- Tanikawa C, Zhang YZ, Yamamoto R, et al. The Transcriptional Landscape of p53 Signalling Pathway. EBioMedicine. 2017;20:109-119.
- Takano Y, Masuda T, Iinuma H, et al. Circulating exosomal microRNA-203 is associated with metastasis possibly via inducing tumor-associated macrophages in colorectal cancer. Oncotarget. 2017;8(45):78598-78613.
- Sato R, Shibata T, Tanaka Y, et al. Requirement of glycosylation machinery in TLR responses revealed by CRISPR/Cas9 screening. Int Immunol. 2017;29(8):347-355.
- Onuki R, Yamaguchi R, Shibuya T, Kanehisa M, Goto S. Revealing phenotype-associated functional differences by genome-wide scan of ancient haplotype blocks. PLoS One. 2017;12(4):e0176530.
- Moriyama T, Shiraishi Y, Chiba K, Yamaguchi R, Imoto S, Miyano S. OVarCall: Bayesian Mutation Calling Method Utilizing Overlapping Paired-End Reads. IEEE Trans Nanobioscience. 2017;16(2):116-122.
- Miyamoto T, Tanikawa C, Yodsurang V, et al. Identification of a p53-repressed gene module in breast cancer cells. Oncotarget. 2017;8(34):55821-55836.
- Jimbo K, Yokoyama K, Ogawa M, et al. [Autologous peripheral blood stem cell transplantation for double-refractory myeloma with K-RAS and N-RAS mutations]. Rinsho Ketsueki. 2017;58(12):2380-2385.
- Ikeda Y, Kiyotani K, Yew PY, et al. Clinical significance of T cell clonality and expression levels of immune-related genes in endometrial cancer. Oncol Rep. 2017;37(5):2603-2610.
- Fujii K, Miyahara Y, Harada N, et al. Identification of an immunogenic neo-epitope encoded by mouse sarcoma using CXCR3 ligand mRNAs as sensors. Oncoimmunology. 2017;6(5):e1306617.
- 2016年
- Yoshino T, Katayama K, Horiba Y, et al. The Difference between the Two Representative Kampo Formulas for Treating Dysmenorrhea: An Observational Study. Evid Based Complement Alternat Med. 2016;2016:3159617.
- Yoshino T, Katayama K, Horiba Y, et al. Predicting Japanese Kampo formulas by analyzing database of medical records: a preliminary observational study. BMC Med Inform Decis Mak. 2016;16:118.
- Yamaguchi K, Nagayama S, Shimizu E, et al. Reduced expression of APC-1B but not APC-1A by the deletion of promoter 1B is responsible for familial adenomatous polyposis. Sci Rep. 2016;6:26011.
- Tamura K, Hazama S, Yamaguchi R, et al. Characterization of the T cell repertoire by deep T cell receptor sequencing in tissues and blood from patients with advanced colorectal cancer. Oncol Lett. 2016;11(6):3643-3649.
- Sugimachi K, Yamaguchi R, Eguchi H, et al. 8q24 Polymorphisms and Diabetes Mellitus Regulate Apolipoprotein A-IV in Colorectal Carcinogenesis. Ann Surg Oncol. 2016;23(Suppl 4):546-551.
- Muramatsu T, Kozaki KI, Imoto S, et al. The hypusine cascade promotes cancer progression and metastasis through the regulation of RhoA in squamous cell carcinoma. Oncogene. 2016;35(40):5304-5316.
- Moriyama T, Shiraishi Y, Chiba K, Yamaguchi R, Imoto S, Miyano S. OVarCall: Bayesian Mutation Calling Method Utilizing Overlapping Paired-End Reads. 2016.
- Kayano M, Matsui H, Yamaguchi R, Imoto S, Miyano S. Gene set differential analysis of time course expression profiles via sparse estimation in functional logistic model with application to time-dependent biomarker detection. Biostatistics. 2016;17(2):235-248.
- Hasegawa T, Niida A, Mori T, et al. A likelihood-free filtering method via approximate Bayesian computation in evaluating biological simulation models. Computational Statistics & Data Analysis. 2016;94:63-74.
- Chapman CG, Yamaguchi R, Tamura K, et al. Characterization of T-cell Receptor Repertoire in Inflamed Tissues of Patients with Crohn's Disease Through Deep Sequencing. Inflamm Bowel Dis. 2016;22(6):1275-1285.
- 2015年
- Yew PY, Alachkar H, Yamaguchi R, et al. Quantitative characterization of T-cell repertoire in allogeneic hematopoietic stem cell transplant recipients. Bone Marrow Transplant. 2015;50(9):1227-1234.
- Yamaguchi K, Komura M, Yamaguchi R, et al. Detection of APC mosaicism by next-generation sequencing in an FAP patient. J Hum Genet. 2015;60(5):227-231.
- Nakata A, Yoshida R, Yamaguchi R, et al. Elevated beta-catenin pathway as a novel target for patients with resistance to EGF receptor targeting drugs. Sci Rep. 2015;5:13076.
- Iwakawa R, Kohno T, Totoki Y, et al. Expression and clinical significance of genes frequently mutated in small cell lung cancers defined by whole exome/RNA sequencing. Carcinogenesis. 2015;36(6):616-621.
- Ikenoue T, Yamaguchi K, Komura M, et al. Attenuated familial adenomatous polyposis with desmoids caused by an APC mutation. Hum Genome Var. 2015;2:15011.
- Hasegawa T, Mori T, Yamaguchi R, et al. Genomic data assimilation using a higher moment filtering technique for restoration of gene regulatory networks. BMC Syst Biol. 2015;9:14.
- Chikahara Y, Niida A, Yamaguchi R, Imoto S, Miyano S. Integrative clustering of cancer genome data using infinite relational models. Proceedings of the 7th International Conference on Bioinformatics and Computational Biology, BICOB 2015. 2015:11-18.
研究キーワード
データ科学、ベイズ統計モデリング、深層学習モデリング、人工知能、データ解析、スーパーコンピュータ、次世代シークエンスデータ、免疫ゲノムデータ、がんゲノム臨床シークエンスデータ情報解析基盤、動的システムモデリング
今後ますます医学・データ科学双方の能力を身に着けた人材が必要とされ活躍の場が広がっています。データ科学の力でがんのシステムの理解を進め、医療・生命科学へ貢献することに興味と熱意のある大学院生を募集しています。