Laboratories
- Back
- Top > Laboratories > Department of Neuroscience > Neurogenetics
Advanced Medical Science(Cooperating field)Neurogenetics
Introduction
(All of the following information is outdated and will be updated in due course.)
This division has started in September 2004 as a new section in the Center for Neurological Diseases and Cancer at the Nagoya University Graduate School of Medicine. Our major research interests include (1) physiology, pathomechanisms, and development of novel therapeutic options for neuromuscular signal transmission and other signal transmissions leading to muscle contractions, (2) molecular mechanisms and their aberrations of RNA processing especially of pre-mRNA splicing, (3) development of novel therapeutic options by exploiting the drug repositioning strategy for neuromuscular and skeletal disorders, (4) roles of intestinal microbiota on development of Parkinson’s disease, and (5) molecular mechanisms of divergent effects of molecular hydrogen on oxidative stress-mediated diseases.
Research Projects
1. Molecular mechanisms and development of therapeutic options for defective neuromuscular signal transmission
i) Congenital myasthenic syndromes
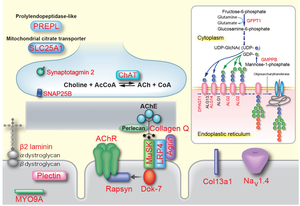
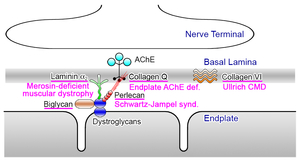
Congenital myasthenic syndromes (CMS) are heterogeneous disorders caused by germline mutations in genes expressed at the neuromuscular junction (Figure 1) (eLS DOI: 10.1002/9780470015902.a0024314, 2014). Mutations have been identified in 24 genes encoding acetylcholine receptor (AChR) subunits (CHRNA1, CHRNB1, CHRND, CHRNE and CHRNG), skeletal muscle sodium channel (SCN4A), signaling molecules driving clustering of AChR (AGRN, LRP4, MUSK and DOK7), synaptic structural proteins (COLQ, LAMB2 and COL13A1), postsynaptic structural proteins (RAPSN and PLEC), presynaptic molecules (CHAT and SYT2), glycosylation enzymes (GFPT1, DPAGT1, ALG2, ALG14 and GMPPB), and other less characterized molecules (PREPL and SCL25A1). CMS are recessive disorders, except for slow channel CMS caused by a missense mutation in one of AChR subunit genes and synaptotagmin 2 (SYT2)-CMS.
Onsets are mostly less than 2 years, but adult-onset is not rare, especially in slow-channel CMS and limb-girdle type CMS caused by glycosylation defects and by DOK7 mutations. Clinical features include fatigable muscle weakness, amyotrophy, and minor facial anomalies. Eye, facial and bulbar muscles are frequently affected, but sparing of these muscles is observed, especially in limb-girdle type CMS. We have first identified molecular defects in (i) choline acetyltransferase that resynthesizes acetylcholine from choline at the nerve terminal (Proc Natl Acad Sci U S A 98: 2017, 2001), (ii) collagen Q (ColQ) that anchors catalytic subunits of acetylcholinesterase to the synaptic basal lamina (Proc Natl Acad Sci U S A 95: 9654, 1998), (iii) AChR that opens cationic ion channel in response to acetylcholine released from the nerve terminal (Proc Natl Acad Sci U S A 92: 758, 1995, Neuron 17: 157, 1996), (iv) rapsyn that clusters AChR at the synaptic basal lamina (Am J Hum Genet 70: 875, 2002), (v) skeletal muscle voltage-gated sodium channel that senses membrane desensitization generated by AChR and opens sodium ion channel (Proc Natl Acad Sci U S A 100: 7377, 2003), and (vi) LRP4 that transmits a signal for clustering of AChR (Hum Mol Genet 23: 1856, 2014, JAMA Neurol 72: 889, 2015).
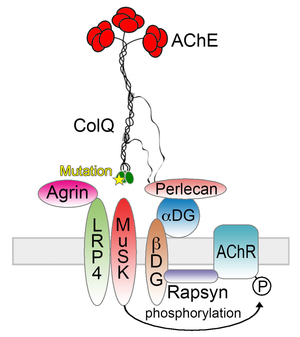
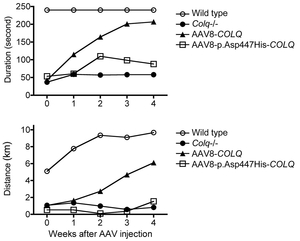
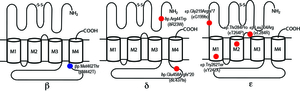
Only a single case of CMS has been genetically diagnosed in Japan before 2009. We started genetic diagnosis of CMS patients mostly using the next generation sequencing techniques and identified mutations in more than 20 patients. We proved that COLQ mutations impair binding of ColQ to MuSK (Figures 2 and 3) (Hum Mutat 34: 997, 2013). We also confirmed that introduction of the mutant ColQ into Colq-knockout mice using the protein-anchoring strategy, which is stated in the next section, does not ameliorate motor deficits (Figure 4). We additionally reported mutations in the AChR subunits with features of slow-channel syndrome and AChR deficiency (Figure 5) (Neuromuscul Disord 25: 60, 2015). Kinetic analysis of another slow-channel mutation in the AChR epsilon subunit gene revealed that a valine ring constituting a channel pore of AChR and comprised of five AChR subunits is critical for stabilizing AChR channel opening (Figure 6) (Hum Mutat 37: 1051, 2016). We are extensively looking for mutations in novel molecules and scrutinizing the underlying molecular pathomechanisms.
 |
 |
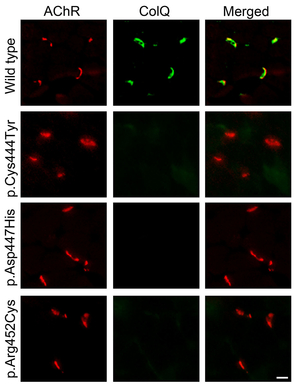 |
| Figure 1. Molecules at the neuromuscular junction that are defective in congenital myasthenic syndromes. | Figure 2. ColQ is anchored to the synaptic basal lamina by binding to MuSK and perlecan. | Figure 3. Mutations in ColQ impair binding of ColQ to the neuromuscular junction of Colq-knockout mice. |
 |
 |
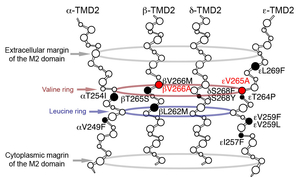 |
| Figure 4. Introduction of mutant D447H-COLQ using AAV8-mediated protein anchoring strategy fails to ameliorate motor deficits of Colq-knockout mice. | Figure 5. Mutations (red symbols) identified in the acetylcholine receptor subunits in Japanese patients with congenital myasthenic syndrome. Single channel analysis revealed that the blue symbol is a normal polymorphism unique to Japanese population. | Figure 6. Valine-to-alanine mutations at the valine ring of the channel pore of the acetylcholine receptor cause slow-channel syndrome. |
ii) Protein-anchoring therapy
Gene therapy can essentially cure any kinds of cells modeling for human diseases. Treatment of human diseases with gene therapy, however, is hindered by unavailability of specific delivery of a transgene to the target organ. An extracellular matrix (ECM) protein carries proprietary binding domain(s), which target the protein to the property site(s). We exploited the unique feature of ECM proteins to ameliorate congenital defects of an ECM protein or to augment expression of an ECM protein.
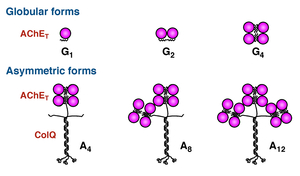
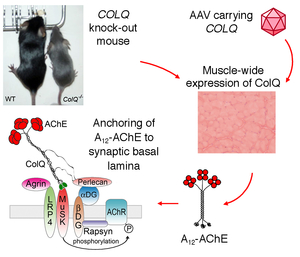
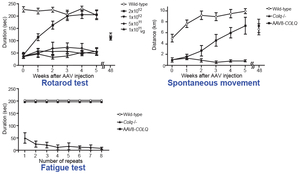
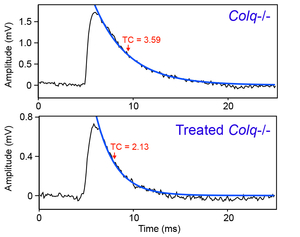
There has been no rational therapy for endplate acetylcholinesterase (AChE) deficiency caused by genetic defects of COLQ encoding collagen Q. Asymmetric AChE species that are composed of AChE and collagen Q (Figure 7) are extracellular matrix molecules that are anchored to the synaptic basal lamina using the collagen domain and the C-terminal domain. We expected that introduction of AAV8-COLQ into skeletal muscles of Colq-knockout mice enables production of asymmetric AChE species, which is secreted from skeletal muscles and anchored to the synaptic basal lamina using its proprietary anchoring domains (Figure 8). We confirmed the feasibility of the protein-anchoring therapy with AAV8-COLQ and observed prominent effects in Colq-deficient mice (Figures 9, 10, and 11) (Mol Ther 20: 1384, 2012). We exploited this strategy to augment expression of biglycan in mdx mice modeling for Duchenne muscular dystrophy, and provided additional evidence that the protein-anchoring strategy can be applied to ECM molecules (Figure 12). (Hum Gene Ther in press).
 |
 |
 |
| Figure 7. Six species of acetylcholinesterase (AChE) expressed in skeletal muscle. | Figure 8. Protein-anchoring therapy for Colq-knockout mice. | Figure 9. Protein-anchoring therapy improved movements of Colq-knockout mice (Fig09_ColQ_movie.mov). |
 |
 |
 |
| Figure 10. Protein-anchoring therapy normalized motor functions of Colq-knockout mice. | Figure 11. Protein-anchoring therapy shortened MEPP decay time. | Figure 12. Extracellular matrix proteins for which the protein-anchoring therapy is potentially effective. |
iii) Identification of novel molecules essential for clustering of acetylcholine receptor (AChR)
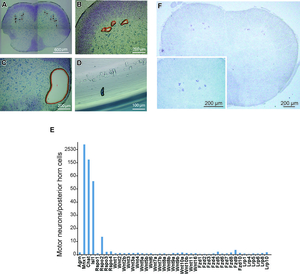
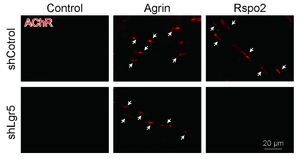
In an effort to search for novel molecules that are essential for clustering of AChR at the neuromuscular junction (NMJ), we performed laser capture microdissection of mouse spinal motor neurons (SMNs) followed by gene expression profilings with microarray and RNA-seq. We found that a secreted activator of Wnt signaling, R-spondin 2 (Rspo2), is highly expressed in SMNs, and is enriched at the neuromuscular junction (Figure 13). Rspo2 induces MuSK phosphorylation and AChR clustering in cultured myotubes in an agrin-independent manner (Figure 14). Leucine-rich repeat-containing G-protein coupled receptor 5 (Lgr5) accumulated at the NMJ is associated with MuSK via LRP4, and serves as a receptor for Rspo2. In Rspo2-knockout mice, the number and density of AChRs at the NMJ are reduced, and the NMJ is ultrastructurally abnormal with widened synaptic clefts and sparse synaptic vesicles (Figure 15) (Sci Rep 6: 28512, 2016). We are also scrutinizing two other novel molecules that drive AChR clustering at the NMJ.
 |
 |
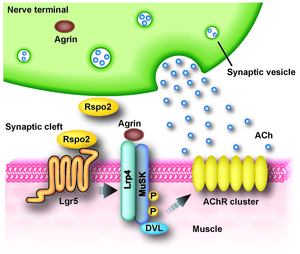 |
| Figure 13. Laser capture microdissection of the mouse spinal motor neurons (SMNs) reveals that Rspo2 is highly expressed in SMNs. | Figure 14. Rspo2 induces clustering of acetylcholine receptor (AChR) in the absence of agrin. Knockdown of the Lgr5 receptor suppresses Rspo2-mediated clustering of AChR. | Figure 15. Schematic of Rspo2-meidated clustering of the acetylcholine receptor. |
iv) Dissection of molecular targets of anti-MuSK antibody
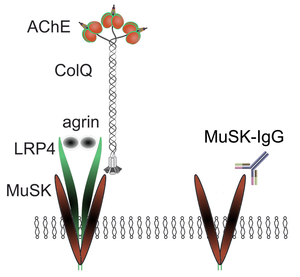
Myasthenia gravis is caused by anti-AChR, anti-MuSK, or anti-LRP4 antibodies. MuSK physiologically binds to LRP4 and ColQ, which constitutes the acetylcholinesterase/ColQ complex. We proved that anti-MuSK antibody blocks binding of ColQ to MuSK in vitro, and causes partial endplate acetylcholinesterase deficiency in mice that were passively transferred with patient’s anti-MuSK antibodies (Figure 16) (Neurology 77: 1819, 2011). We later proved that anti-MuSK antibody also blocks binding of MuSK to LRP4 in vitro in the presence of agrin. Passive transfer of anti-MuSK antibody to Colq-knockout mice similarly attenuated AChR clustering, indicating that AChR deficiency in anti-MuSK patients is likely due to blocking of LRP4, not of ColQ (Sci Rep 5: 13928, 2015). To our surprise, binding of ColQ to MuSK physiologically blocks MuSK-LRP4 interaction and suppresses agrin/LRP4/MuSK signaling. Quantitative analysis revealed that anti-MuSK antibody suppresses agrin/LRP4/MuSK signaling to a greater extent than the AChE/ColQ complex (Figure 17). This was why replacement of ColQ by anti-MuSK antibody attenuates agrin/LRP4/MuSK signaling. We are currently further dissecting mutual interactions between agrin, LRP4, MuSK, ColQ, biglycan, and anti-MuSK antibodies.
 |
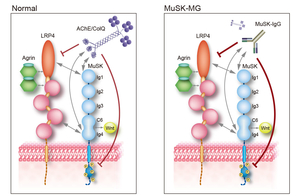 |
| Figure 16. Anti-MuSK antibody blocks binding of ColQ to MuSK. | Figure 17. Acetylcholinesterase (AChE)/ColQ complex blocks binding of MuSK to LRP4 and suppresses MuSK phosphorylation. Anti-MuSK antibody displaces AChE/ColQ and further blocks binding of MuSK to LRP4, and inhibits MuSK phosphorylation. |
2. Physiological and pathological regulations of RNA metabolisms
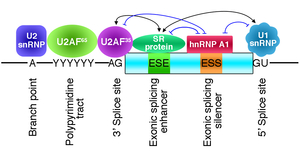
i) Disclosure of hidden scenarios of splicing cis-elements, and development of tools to predict splicing consequences of mutations affecting splicing cis-elements
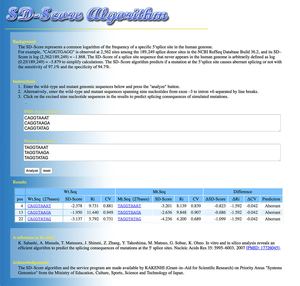
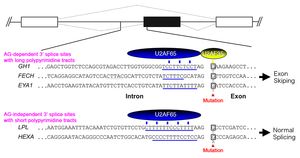
We humans exploit tissue-specific and developmental stage-specific alternative splicing to express more than 100,000 proteins from ~18,000 genes. More than 96% of human multi-exon genes are known to be alternatively spliced. Alternative splicing is achieved by splicing cis-elements on each gene and splicing trans-factors that are regulated in tissue-specific and developmental stage-specific manners (Figure 18). Classical splicing cis-elements include the branch point sequence, the polypyrimidine tract, and the 3’ and 5’ splice sites. These cis-elements are highly degenerative. The degeneracy makes prediction of splicing consequence of mutations that disrupt splicing cis-elements difficult and equivocal. We developed an algorithm that we named the SD Score to predict if a given mutation at the 5’ splice site affects pre-mRNA splicing or not. The SD Score algorithm is available at our web server (Figure 19) (Nucleic Acids Res 35: 5995, 2007). The human branch point consensus sequences had been simply derived from similarity to the highly conserved yeast branch point sequence of UACUAAC, which makes prediction of splicing consequences of branch point-disrupting mutations difficult. We have experimentally disclosed that the human branch point consensus sequence is yUnAy (Figure 20) (Nucleic Acids Res 36: 2257, 2008). In addition, a single nucleotide substitution at the first nucleotide of an exon causes aberrant splicing only when the polypyrimidine tract in the preceding intron is short (an AG-dependent 3’ splice site) (Figure 21) (Nucleic Acids Res 39: 4396, 2011). We have also developed IntSplice, which predicts splicing consequences of mutations from positions -50 to -3 from the 3’ end of an intron including the branch point sequence and the polypyrimidine tract (Figure 22) (J Hum Genet 2016). IntSplice is available on our web server. Only a few researchers in the world are interested in predicting the splicing effects of mutations. We are currently developing similar tools for mutations at the other sites.
Using similar techniques, we are developing a tool to predict pathogenicity of amino acid substitutions. Currently, the ROC curve of our tool is better than any other 24 previously reported evaluation tools.
 |
 |
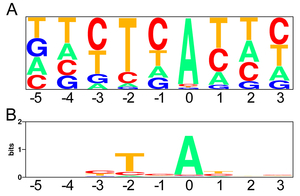 |
| Figure 18. Splicing cis-elements and trans-factors | Figure 19. SD-Score algorithm predicts the splicing consequence of a mutation affecting the 5’ splice site with 97.1% sensitivity and 94.7% specificity | Figure 20. Pictogram (A) and WebLogo (B) of human branch point consensus sequence |
 |
 |
| Figure 21. Mutations at the first nucleotide of an exon at AG-dependent 3’ splice sites with short polypyrimidine tracts cause aberrant splicing | Figure 22. IntSplice web server predicts the splicing consequence of a mutation at intronic position -50 to -3 close to the 3’ end of an intron. |
ii) Molecular mechanisms underlying disruption of splicing cis-elements in human diseases
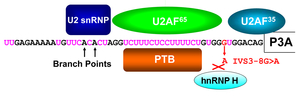
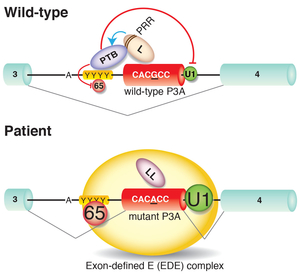
We dissect molecular mechanisms underlying exonic and intronic splicing mutations that do not affect conventional splicing cis-elements. An intronic mutation in CHRNA1 encoding the AChR alpha subunit identified in a patient with CMS disrupts a splicing cis-element and decreases a binding affinity for hnRNP H (Figure 23) (Hum Mol Genet 17: 4022, 2008). We also proved that the polypyrimidine tract binding protein (PTB) also binds to a neighboring site (Hum Mol Genet 18: 1229, 2009). Another exonic mutation in another CMS patient disrupts binding of hnRNP L and de novo gains binding of hnRNP LL (Figure 24) (Sci Rep 3: 2931, 2013). Similarly, a mutation disrupting a splicing enhancing cis-element on exon 16 of COLQ encoding collagen Q, loses binding of SRSF1 and de novo gains binding of hnRNP H, which cause exclusive skipping of COLQ exon 16 (Figure 25) (Sci Rep 5: 13208, 2015). We are currently working on additional mutations disrupting other splicing cis-elements in congenital myasthenic syndromes and other diseases.
 |
 |
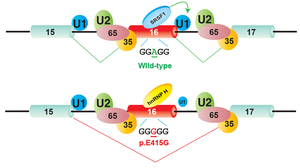 |
| Figure 23. CHRNA1 exon P3A is suppressed by hnRNP H and PTB. A mutation in a patient attenuates binding of hnRNP H and provokes exon recognition. | Figure 24. Proline-rich region (PRR) of hnRNP L (L) binds to PTB and suppresses exon P3A of CHRNA1. A G-to-A mutation in a patient displaces hnRNP L and de novo gains binding of hnRNP LL (LL) that lacks PRR, and provokes exon recognition. | Figure 25. Constitutive COLQ exon 16 is enhanced by SRSF1. A mutation disrupts binding of SRSF1, and de novo gains binding of hnRNP H, and causes exon skipping. |
iii) Molecular mechanisms of alternative splicing of genes expressed at the neuromuscular junction (NMJ)
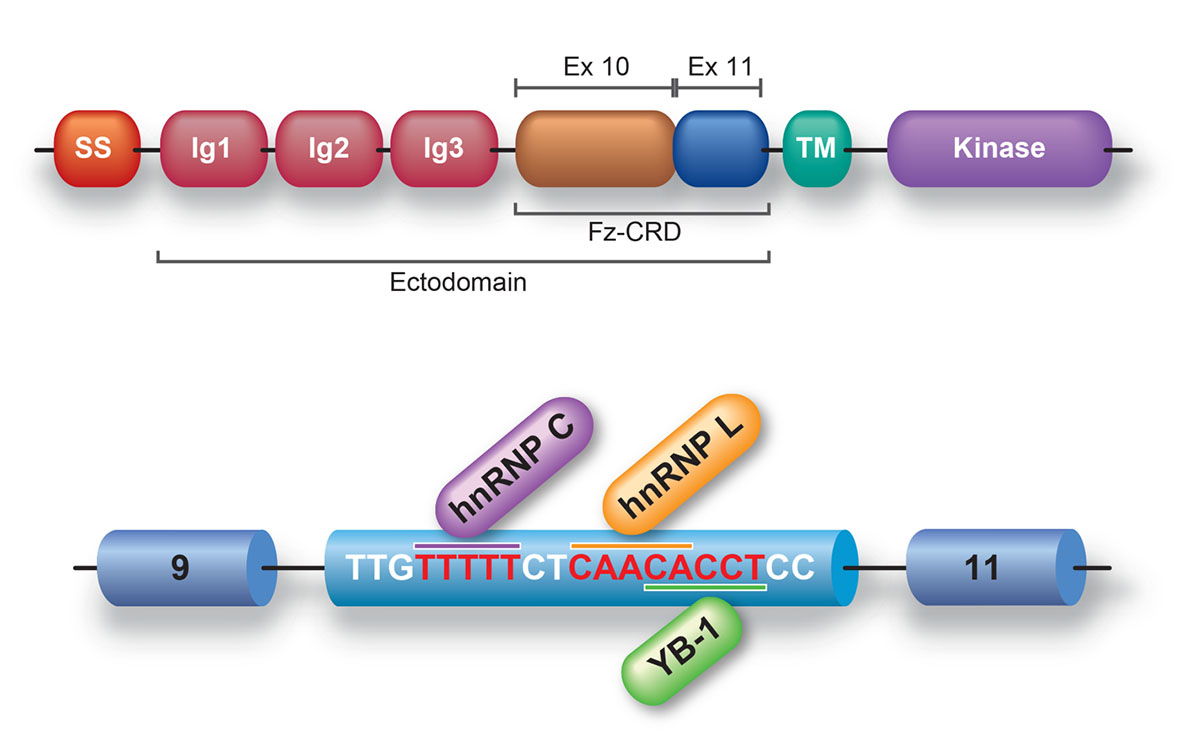
MUSK exon 10 is alternatively skipped in human, but not in mouse. Skipping of exon 10 disrupts a cysteine-rich region (Fz-CRD), which is essential for Wnt-mediated AChR clustering (Figure 26). Block-scanning mutagenesis, RNA-affinity purification, mass spectrometry, Western blotting, siRNA-mediated gene knockdown revealed that hnRNP C, YB-1 and hnRNP L bind to MUSK exon 10 and cause skipping of exon 10. Antibody-mediated in vitro protein depletion and scanning mutagenesis additionally revealed that hnRNP C promotes binding of YB-1 and hnRNP L to the immediate downstream sites and enhances exon skipping. Simultaneous tethering of two splicing trans-factors to the target confirmed the cooperative effect of YB-1 and hnRNP L on hnRNP C-mediated exon skipping. Search for a similar motif in the human genome revealed nine alternative exons that were individually or coordinately regulated by hnRNP C and YB-1 (Sci Rep 4: 6841, 2014).
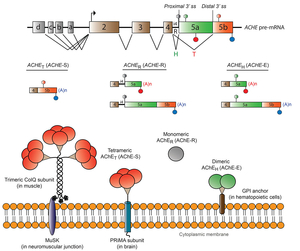
Acetylcholinesterase (AChE), encoded by the ACHE gene, hydrolyzes the neurotransmitter acetylcholine to terminate synaptic transmission. Alternative splicing close to the 3' end generates three distinct isoforms of AChE-T, AChE-H and AChE-R (Figure 27). We found that hnRNP H binds to two specific G-runs in exon 5a of human ACHE and activates the distal alternative 3' splice site (ss) between exons 5a and 5b to generate AChET (Figure 28). Specific effect of hnRNP H was corroborated by siRNA-mediated knockdown and artificial tethering of hnRNP H. Furthermore, hnRNP H competes for binding of CstF64 to the overlapping binding sites in exon 5a, and suppresses the selection of a cryptic polyadenylation site (PAS), which additionally ensures transcription of the distal 3' ss required for the generation of AChE-T. Expression levels of hnRNP H were positively correlated with the proportions of the AChE-T isoform in three different cell lines. HnRNP H thus critically generates AChE-T by enhancing the distal 3' ss and by suppressing the cryptic PAS. Global analysis of CLIP-seq and RNA-seq also revealed that hnRNP H competitively regulates alternative 3' ss and alternative PAS in other genes. HnRNP H and CstF64 play pivotal roles in switching alternative splicing and alternative polyadenylation (Nucleic Acids Res, in press).
We are dissecting molecular mechanisms of more genes expressed at the neuromuscular junction.
 |
 |
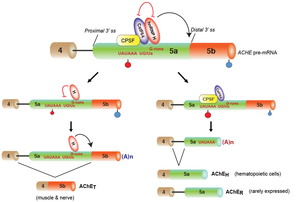 |
| Figure 26. Fz-CRD of MuSK is encoded by exons 10 and 11. MUSK exon 10 is alternatively skipped. Binding of hnRNP C to exon 10 enhances binding of hnRNP L and YB-1 to its immediate downstream, and coordinately enhances skipping of exon 10. | Figure 27. Schematic of alternative splicing of AChE isoforms and their membrane localizations. | Figure 28. Schematic showing competition between hnRNP H-mediated activation of the distal 3’ ss generating AChET and CstF64-mediated activation of the cryptic polyadenylation site (UAUAAA) generating AChE-H and AChE-R. The cryptic (pA-1) and canonical (pA-2) PASs are marked by red and blue closed circles, respectively. HnRNP H-binding G-runs and CstF64-binding UG/Us are overlapping, and hnRNP H and CstF64 compete for binding to exon 5a. |
iv) Extensive splicing analysis with exon array and RNA-seq
We use both Affymetrix exon array and RNA-seq to detect alternative and aberrant splicing events. In an effort to identify aberrant splicing events in myotonic dystrophy (DM1), we developed an analysis tool for the Affymetrix exon array. Extensive analysis of different parameters disclosed that Z-score of each exon best discriminates false- and true-positives (Figure 29) (J Hum Genet 57: 368, 2012).
Comparison of the exon array and RNA-seq currently leads to a tentative conclusion that exon array is better than RNA-seq for analyzing expression levels, which is likely because PCR is included in RNA-seq, but not in the exon array.
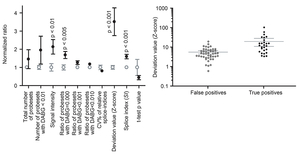 |
| Figure 29. Deviation value (Z-score) best discriminates false- and true-positives of the Affymetrix exon array. |
v) Elucidation of the roles of RNA-binding proteins by multi-OMICS analysis including CLIP-seq
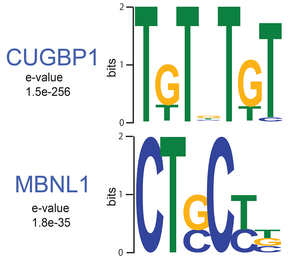
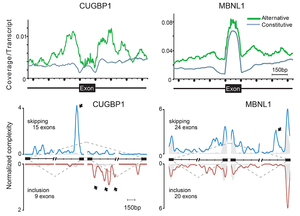
MBNL1 and CUGBP1 are RNA-binding proteins that are misregulated in myotonic dystrophy (DM1). We determined RNA targets recognized by MBNL1 and CUGBP1 by CLIP-seq. We identified binding motifs (Figure 30) and position-specific splicing regulations of both proteins (Figure 31) (Sci Rep 2: 209, 2012). In addition, we found that both proteins preferentially bind to 3’ UTR and facilitate degradation of target transcripts (Figure 32). We additionally analyzed RNA targets of FUS by CLIP-seq, which is misregulated in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). We found that FUS binds to non-coding RNA arising from the antisense strands in the promoter regions and downregulates transcriptions of the target genes (Figure 33) (Sci Rep 2: 529, 2012). Multi-OMICS analyses using CLIP-seq, ChIP-seq, RNA-seq, CAGE-seq, and polyA-seq along with molecular dissections of individual FUS-regulated genes revealed that binding of FUS to nascent RNA stalls RNA polymerase II. If FUS binds immediately downstream to an alternative polyadenylation site, CPSF160 is recruited to the nascent RNA and polyadenylated short mRNA is generated, which also reduces the amount of polyadenylated long mRNA. If FUS binds immediately upstream to an alternative polyadenylation site, CPSF160 is not recruited to nascent RNA and the short nascent RNA transcript is degraded (Figure 34) (Genes Dev 29: 1045, 2015). We are developing a novel CLIP-seq technique to unveil yet unknown functions of RNA-binding proteins in a small scale.
 |
 |
 |
| Figure 30. RNA-binding motifs of CUGBP1 and MBNL1. | Figure 31. Position-specific splicing regulation by CUGBP1 and MBNL1. | Figure 32. CUGBP1 and MBNL1 bind to 3’ UTRs and facilitate mRNA degradations. |
 |
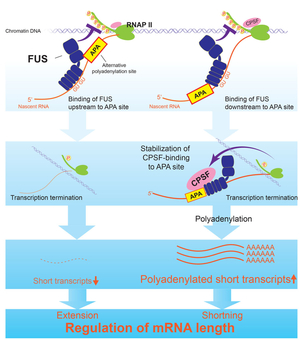 |
| Figure 33. FUS binds to non-coding RNA derived from an antisense strand in the promoter region. | Figure 34. Position-specific binding of FUS to nascent RNA regulates mRNA length. |
vi) siRNA-designing algorithm
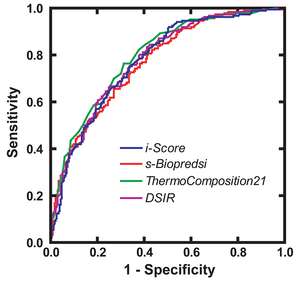
RNA interference is commonly used as a modality for straightforward and robust suppression of the target gene. We applied our modeling techniques for designing siRNA, and developed an algorithm to design efficient siRNAs that we named iScore (Figures 35 and 36) (Nucleic Acids Res 35: e123, 2007). We have a web service program, iScore, which simultaneously calculates eight different siRNA-designing scores including iScore (Figure 37). Referring to the eight scores, we routinely make two siRNAs, and on average one of the two siRNAs suppresses the gene expression level down to more than 20%.
 |
 |
 |
| Figure35.Sensitivity and specificity of the iScore siRNA-designing algorithm are comparable to those of three second-generation designing algorithms. | Figure36.Heat-unstable siRNAs demonstrate an efficient ROC curve. | Figure37. iScore designer web service program. |
3. Drug repositioning strategy for orphan diseases
i) Repositioning of pre-approved drugs for neuromuscular disorders
Pre-approved drugs exhibit unpredicted beneficial effects on totally unrelated disorders, as observed in the anti-platelet effect of aspirin (NSAID); an anti-tremor effect of propranolol (adrenergic blocker); an anti-tumor effect of thalidomide (hypnotics); and anti-Parkinsonian effects of amantadine (anti-viral agent) and zonisamide (anti-epileptics) to name a few. We know optimal and maximal dosages, optimal routes and protocols of drug administrations, safety margins, and potential adverse effects of pre-approved drugs. With these advantages, information that we obtained in cultured cells and model animals can be readily applied to the clinical settings.
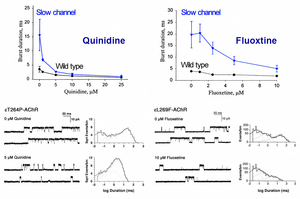
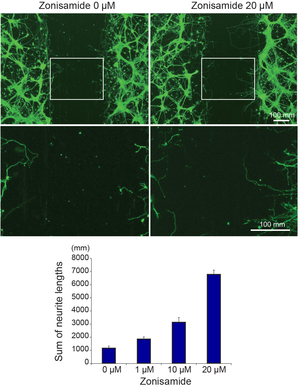
Slow channel syndrome is caused by prolonged opening episodes of the acetylcholine receptor. We have previously shown that anti-arrhythmic agent, quinidine (Ann Neurol 43: 480, 1998), and anti-depressant, fluoxetine (Neurology 60: 1710, 2003), ameliorate abnormally prolonged opening episodes of the acetylcholine receptor (Figure 38). Screening of neurite elongation of NSC34 cells revealed that an anti-epileptic and anti-Parkinsonian agent, zonisamide, enhances neurite elongation (Figure 39). We confirmed a dose-dependent effect using primary spinal motor neurons isolated from mice. Zonisamide markedly enhances axonal regeneration after an autograft operation of the sciatic nerve in mice (Figure 40) (PLoS One 10: e0142786, 2015). Zonisamide also exhibits effects on a mouse model of congenital myasthenic syndrome.
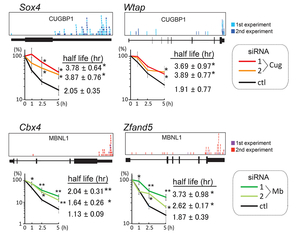
Myotonic dystrophy type 1 (DM1) is caused by abnormal expansion of CTG repeats in the 3’ untranslated region of the DMPK gene. Expanded CTG repeats are transcribed into RNA and make an aggregate with a splicing regulator, MBNL1, in the nucleus, which is called the nuclear foci. A nonsteroidal anti-inflammatory drug (NSAID), phenylbutazone (PBZ), upregulates the expression of MBNL1 in C2C12 mouse myoblasts as well as in a mouse model for DM1. In the DM1 mouse model, PBZ ameliorates aberrant splicing of Clcn1, Nfix, and Rpn2; increases expression of skeletal muscle chloride channel; decreases abnormal central nuclei of muscle fibers; and improves wheel-running activity. The effect of PBZ is attributed to two distinct mechanisms (Figure 41). First, PBZ suppresses methylation of an enhancer region in Mbnl1 intron 1, and enhances transcription of Mbnl1 mRNA. Second, PBZ attenuates binding of MBNL1 to abnormally expanded CUG repeats in cellulo and in vitro (Sci Rep 6: 25317, 2016). As PBZ is an old drug with unignorable adverse effects, we are currently looking for another pre-approved drug that exhibit similar effects.
 |
 |
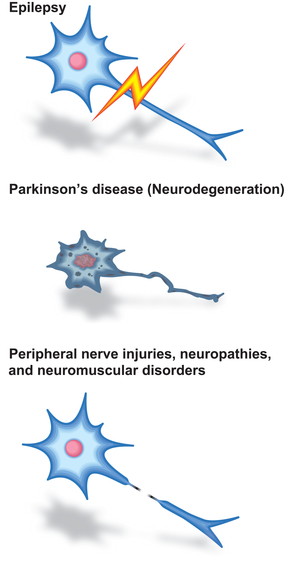 |
| Figure 38. Anti-arrhythmic agent, quinidine, and anti-depressant, fluoxetine, normalize abnormally prolonged ion channel openings in slow channel syndrome. | Figure 39. Zonisamide induces neurite regeneration in the neurite scratch assay of mouse primary spinal motor neurons. | Figure 40. Zonisamide is developed for epilepsy, and repositioned for Parkinson’s disease. Zonisamide can be repositioned again for peripheral nerve injuries, neuropathies, and neuromuscular disorders. |
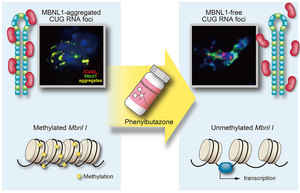 |
| Figure 41. Phenylbutazone improves motor deficits in a mouse model for myotonic dystrophy type 1 by decreasing biding of MBNL1 to the abnormally expanded CUG repeats and by enhancing demethylation of Mbnl1 intron 1 followed by increased expression of Mbnl1. |
ii) Repositioning of pre-approved drugs for skeletal disorders
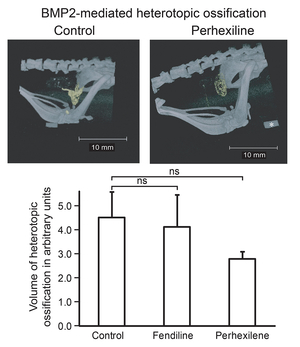
In fibrodysplasia ossificans progressiva (FOP), we found that clinically applied Ca channel blockers, perhexiline and fendiline, are effective for model cells and model mice (Figure 42) (J Bone Miner Metab 31: 26, 2013). An open label trial with perhexiline in five FOP patients, however, showed no beneficial effects (Orphanet J Rare Dis 8: 163, 2013). We are looking for another pre-approved compound to treat FOP.
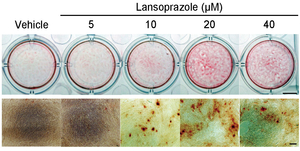
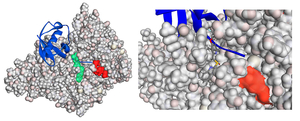
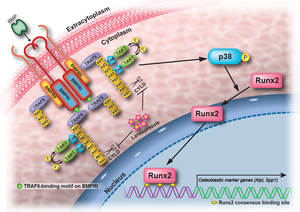
A proton pump inhibitor, lansoprazole, enhances nuclear accumulation of Runx2 and induces osteoblastogenesis (Figure 43). Systemic administration of lansoprazole to a rat femoral fracture model increased osteoblastogenesis. Dissection of signaling pathways revealed that lansoprazole activates a noncanonical bone morphogenic protein (BMP)-transforming growth factor-beta (TGF-beta) activated kinase-1 (TAK1)-p38 mitogen-activated protein kinase (MAPK) pathway. We found by in cellulo ubiquitination studies that lansoprazole enhances polyubiquitination of the TNF receptor-associated factor 6 (TRAF6) and by in vitro ubiquitination studies that the enhanced polyubiquitination of TRAF6 is attributed to the blocking of a deubiquitination enzyme, cylindromatosis (CYLD). Structural modeling and site-directed mutagenesis of CYLD demonstrated that lansoprazole tightly fits in a pocket of CYLD where the C-terminal tail of ubiquitin lies (Figure 44). Lansoprazole is a potential therapeutic agent for enhancing osteoblastic differentiation (Figure 45) (EBioMedicine 2: 2046, 2015).
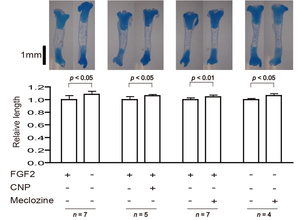
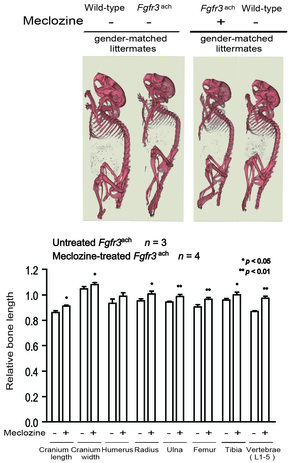
Achondroplasia (ACH) is caused by gain-of-function mutations in FGFR3 encoding the fibroblast growth factor receptor 3. An anti-histamine and anti-emetic agent, meclozine, facilitates chondrocyte proliferation and mitigates loss of extracellular matrix in FGF2-treated rat chondrosarcoma (RCS) cells. Meclozine enhances proliferation of cultured cells modeling for ACH and embryonic tibial bone also modeling for ACH in explant culture (Figure 46) (PLoS One 8: e81569, 2013), as well as ACH model mice (Figure 47) (Endocrinology 156: 548, 2015). A Ca channel blocker, verapamil, induces expression of FRZB, a soluble antagonist of Wnt signaling in human osteoarthritis (OA) chondrocytes. Intraarticular injection of verapamil inhibits OA progression as well as nuclear localization of beta-catenin in a rat OA model (Figure 48) (PLoS One 9: e92699, 2014)
We are currently seeking for more pre-approved agents to treat skeletal disorders.
 |
 |
 |
| Figure 42. Clinically applicable Ca channel blockers, perhexiline and fendiline, suppress heterotopic ossification in FOP model mice | Figure 43. Lansoprazole increases matrix calcium deposition by Alizarin red staining in human bone marrow-derived mesenchymal cells that are induced to differentiate into osteoblasts. | Figure 44. Simulated crystal structure of CYLD (PDB id: 2VHF) bound to ubiquitin (blue) (PDB id: 1NBF). The C-terminal tail of ubiquitin extends to the active site (red). Lansoprazole (green) fits into a pocket to cross the C-terminal tail of ubiquitin. Artificial mutations of R758 (not shown) and F766 (not shown) constituting the pocket to alanine have no effect on the enzymatic activity of CYLD, but nullify the effect of lansoprazole. |
 |
 |
 |
| Figure 45. Proposed model of lansoprazole-induced activation of Runx2. | Figure 46. Meclozine increases the longitudinal length of embryonic tibiae with or without FGF2 treatment in bone explant culture. | Figure 47. Meclozine increases the longitudinal bone growth of Fgfr3ach mice. |
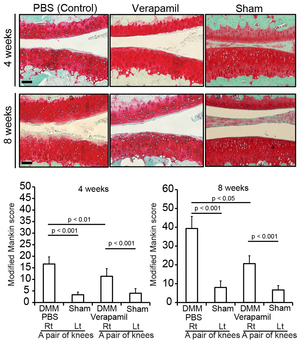 |
| Figure 48. Intraarticular injection of verapamil ameliorates progression of osteoarthritis in a rat model. |
4. Intestinal microbiota in Parkinson’s disease
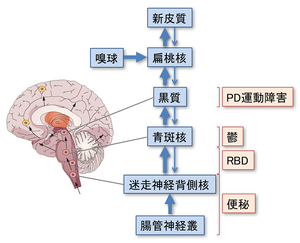
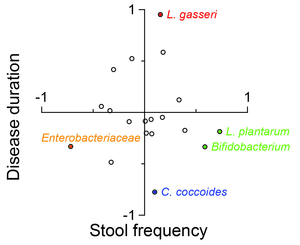
Parkinson’s disease (PD) is characterized by abnormal aggregation of alpha-synuclein in substantia nigra and the other brain regions. It is currently established that abnormal accumulation of alpha-synuclein starts from the intestinal neural plexus and propagates into the dorsal nucleus of vagus nerve followed by locus ceruleus, and substantia nigra (Figure 49). In PD, intestinal permeability is increased and abnormal staining for E. coli and nitrotyrosine is observed in the intestinal wall. In an effort to search for the effects of intestinal microbiota in development of PD, we analyzed intestinal microbiota in 52 PD patients and 36 healthy cohabitants, and observed alterations in microbiota in PD. Modeling analysis revealed that Lactobacillus gasseri is elevated in advanced PD, and Clostridium coccoides is elevated in early PD (Figure 50). In addition, the number of hydrogen-producing bacteria is lower in PD patients compared to controls (Figure 51) (PLoS One 10: e0142164, 2015). We are currently recruiting more PD patients and healthy cohabitants, and analyzing intestinal microbiota using shotgun metagenome analysis and other biochemical markers.
 |
 |
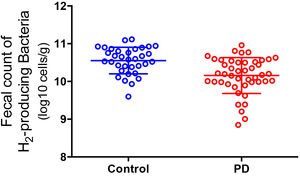 |
| Figure 49. Braak’s observation showing that abnormal accumulation of alpha-synuclein ascends from dorsal nucleus of vagus up to substantia nigra. RBD, REM behavior disorder. | Figure 50. Modeling analysis of gut microbiota in Parkinson’s disease reveals bacteria associated with progression of Parkinson’s disease and with severity of constipation. | Figure 51. The number of hydrogen-producing bacteria is lower in Parkinson’s disease. |
5. Elucidation of molecular mechanisms of the effects of molecular hydrogen on a plethora of diseases and disease models
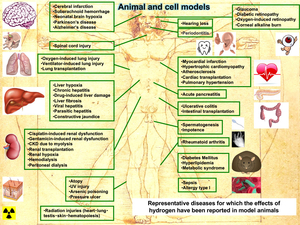
Molecular hydrogen is effective for a wide range of disease models (Figure 52) and human diseases (Figure 53) (Oxid Med Cell Longev 2012: 353152, 2012). A total of 321 original articles have been published from 2007 to 2015 (Med Gas Res 5: 12, 2015). About three-quarters of the articles show the effects in mice and rats. The number of clinical trials is increasing every year. The effects have been reported in essentially all organs covering 31 disease categories that can be subdivided into 166 disease models, human diseases, treatment-associated pathologies, and pathophysiological conditions of plants with a predominance of oxidative stress-mediated diseases and inflammatory diseases. We reported the effects of hydrogen in a rat model of Parkinson’s disease (Figures 54 and 55) (Neurosci Lett 453: 81, 2009), patients with inflammatory and mitochondrial myopathies (Med Gas Res 1: 24, 2011), rat fetal hippocampal damage caused by in utero ischemia-reperfusion (Free Radic Biol Med 69: 324, 2014), pulmonary hypertension in rats (J Thorac Cardiovasc Surg 150: 645, 2015), mdx mice modeling for Duchenne muscular dystrophy (Redox Rep 1, 2016), perinatal brain injury caused by in utero prenatal inflammation (Free Radic Biol Med 91: 154, 2016), and bronchopulmonary dysplasia (BPD) in newborn rats (Pediatr Pulmonol 51: 928, 2016).
Specific extinction of hydroxyl radical and peroxynitrite was initially presented, but the radical-scavenging effect of hydrogen cannot be held solely accountable for its drastic effects. Molecular hydrogen suppresses FcepsilonRI-mediated signal transduction of mast cells (Biochem Biophys Res Commun 389: 651, 2009), and also inhibits lipopolysaccharide/interferon gamma-induced nitric oxide production through modulation of signal transduction in macrophages (Biochem Biophys Res Commun 411: 143, 2011). Extensive analyses of gene expression profiles also demonstrate that hydrogen modulates multiple signaling pathways (Mol Cell Biochem 403: 231, 2015). We also reported that hydrogen suppresses activated Wnt/beta-catenin signaling by enhancing the activity of the degradation complex of beta-catenin (Sci Rep 6: 31986, 2016). In addition, hydrogen water and intermittent hydrogen gas exposure, but not continuous hydrogen gas exposure, prevent 6-hydorxydopamine-induced Parkinson's disease in rats (Med Gas Res 2: 15, 2012). We are currently narrowing down to an exact target of hydrogen, which ignites activation and suppression of a wide range of signaling cascades.
 |
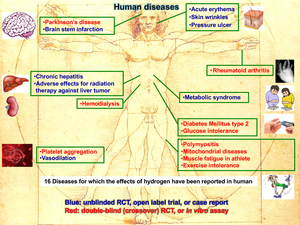 |
| Figure 52. Representative disease models for which molecular hydrogen exhibits beneficial effects. | Figure 53. Human diseases for which molecular hydrogen exhibits beneficial effects. |
|
|
|
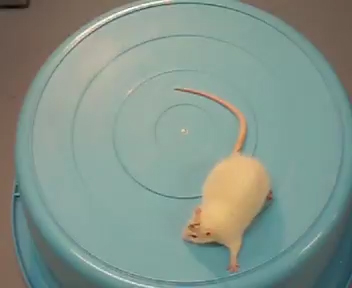
| Figure 54. A rat with hemi-Parkinsonism taking control water vigorously rotates clockwise after intraperitoneal injection of amphetamine (Fig54_Hydrogen_Ctr_Low.mov). | Figure 55. A rat with hemi-Parkinsonism taking hydrogen-rich water does not rotate after intraperitoneal injection of amphetamine (Fig55_Hydrogen_PreH_Low.mov). |

Faculty Members
| Faculty | Position | Department |
|---|---|---|
| Yuka Nakazawa | Professor | Neurogenetics |
| Akio Masuda | Associate Professor | Neurogenetics |
| Mikako Ito | Lecturer | Neurogenetics |
| Tomonari Hamaguchi | Designated Lecturer | Medical and Healthcare Innovation Unit |
Bibliography
- 1988-2024(Same as Japanese page)
- https://www.med.nagoya-u.ac.jp/medical_J/laboratory/neurological-diseases-cancer/dept-neuroscience/neurogenetics/
Research Keywords
Congenital myasthenic syndromes, neuromuscular junction, pre-mRNA splicing, RNA-binding proteins, drug repositioning, Parkinson's disease, intestinal microbiota, molecular hydrogen
Private HP (in Japanese) (inactive)
Call for Graduate Students
We are not taking new students now.