- TOP
- >
- 研究内容
研究内容
当研究室では、生命現象の根幹である遺伝情報の継承および発現制御機構の解明から、がんにおける治療標的同定と治療戦略開発に至るまで統合的な研究を行っています。こちらのページでは主要な研究テーマである「がんの治療標的に関する研究」「発がんに寄与するゲノム安定性維持機構解明」「適切な細胞増殖に必要なチェックポイント機構の提唱と分子機構解明」についてご紹介いたします。
(リファレンスをクリックすると元論文のページへ飛びます。)
がんの治療標的
に関する研究
難治性がんにおける治療標的の同定と治療戦略の開発
乳がんは日本人女性が罹患する悪性腫瘍の第1位であり、その罹患数ならびに死亡数は年々増加しています。女性ホルモンの一種であるエストロゲンと結合する受容体(ERα)が発現している乳がん(ERα陽性乳がん)は、乳がんの約70%を占めています。ERα陽性乳がんに対しては、ERαの働きを抑制する内分泌療法が奏効します。しかしながら、ERα陽性転移乳がん患者に内分泌療法を行った場合、治療当初には内分泌療法に効果を認めても、いずれ内分泌療法に対して耐性を示してしまいます。そのため、内分泌療法に対する耐性メカニズムの解明とその克服が大きな臨床的課題となっています。そこで、乳がんの新たな治療標的を同定するために、①ERαと相互作用する、②ERαと発現量が正の相関関係を示す、③乳がんで過剰発現する、④高発現は生存期間が短縮する、という4つの基準を満たす乳がんの予後不良因子として、FKBP52 (FK506 Binding Protein 52)を同定しました(Fig.1)(Habara et al.,PNAS, 2022)
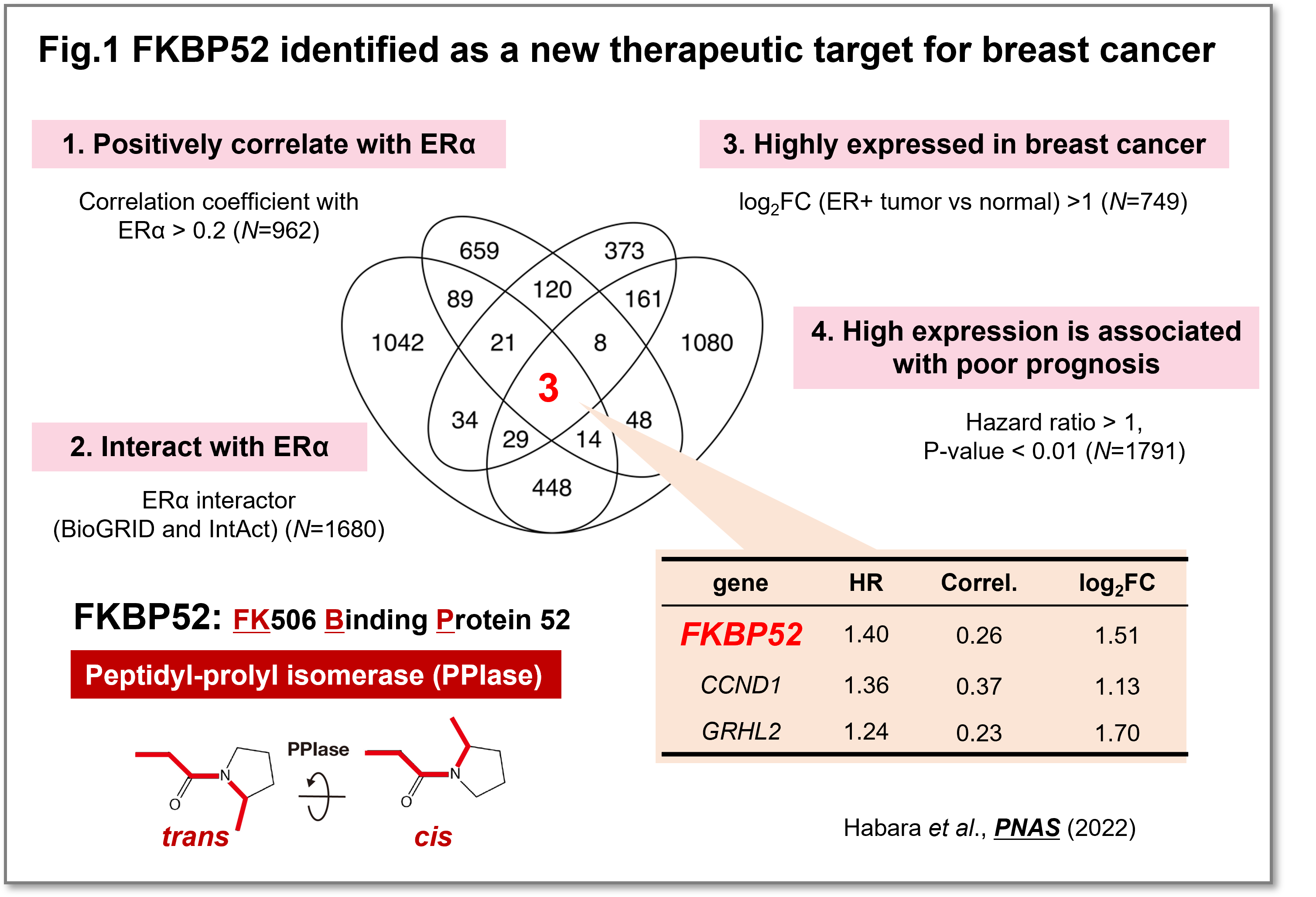
(画像をクリックで拡大)
乳がん細胞株(MCF7)において、FKBP52の発現を抑制すると、ERαの分解が亢進するためERαの発現量が減少し、がん細胞の増殖を顕著に阻害しました。重要なことに、内分泌治療抵抗性となった乳がん細胞株に対しても、FKBP52を阻害することで、ERαの発現量およびがん細胞の増殖を抑制できる結果が得られました。さらにFKBP52はBRCA1とERαの結合を促進することで、ERαを安定化することを解明しました。FKBP52と相同性の高いFKBP51の機能を調べたところ、FKBP51は①乳がんで発現が減少する、②高発現は生存期間を延長する、③ERαの分解を促進するという、FKBP52とは逆の機能を有することが判明しました。以上の結果から、FKBP52はERαを安定化することにより、ERαの機能を増強し、がん細胞の増殖を促進させることが分かりました。一方、FKBP51はFKBP52と競合してERαと結合し、ERαの分解を促進する機能を持つことが示唆されました(Fig.2)。FKBP52阻害は、再発性乳がんに対しても効果があることから、新たなバイオマーカー、治療法開発につながることが期待できます。
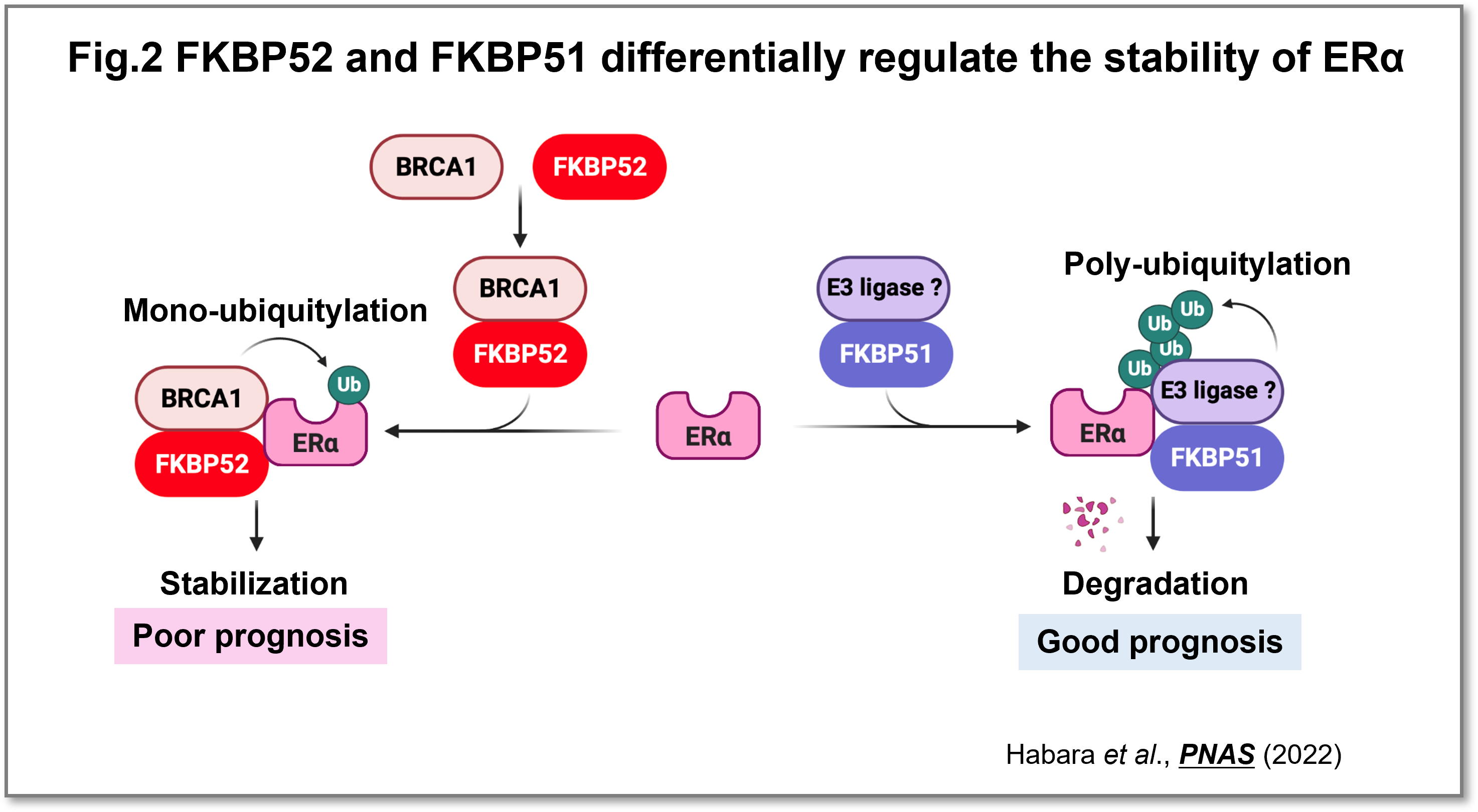
(画像をクリックで拡大)
前立腺がんの悪性化に関わるAR(Androgen receptor)は、細胞内でFKBP51およびFKBP52と複合体を形成しますが、FKBP51, FKBP52によるARの機能調節機能についてはよく理解されていませんでした。私達の解析からFKBP51, FKBP52は前立腺がん細胞の増殖に必須であること、AR標的遺伝子発現に重要であること、リガンド添加時に誘導されるARの二量体形成に重要であることが分かりました(Fig.3)。共同研究によりFKBP51の酵素活性阻害剤を合成することに成功し、酵素活性がARの二量体化と増殖に必要であることが明らかとなりました(Maeda et al.,Mol. Oncol., 2022)。ホルモン療法に対して抵抗性になった去勢抵抗性前立腺がんに対してもFKBP51, FKBP52の阻害が有効であることから、新たな治療選択になりうる可能性が期待できます。
現在、難治性乳がん、前立腺がん以外にもがんの病態と関連する機能を解明し、阻害剤開発へと展開しています。
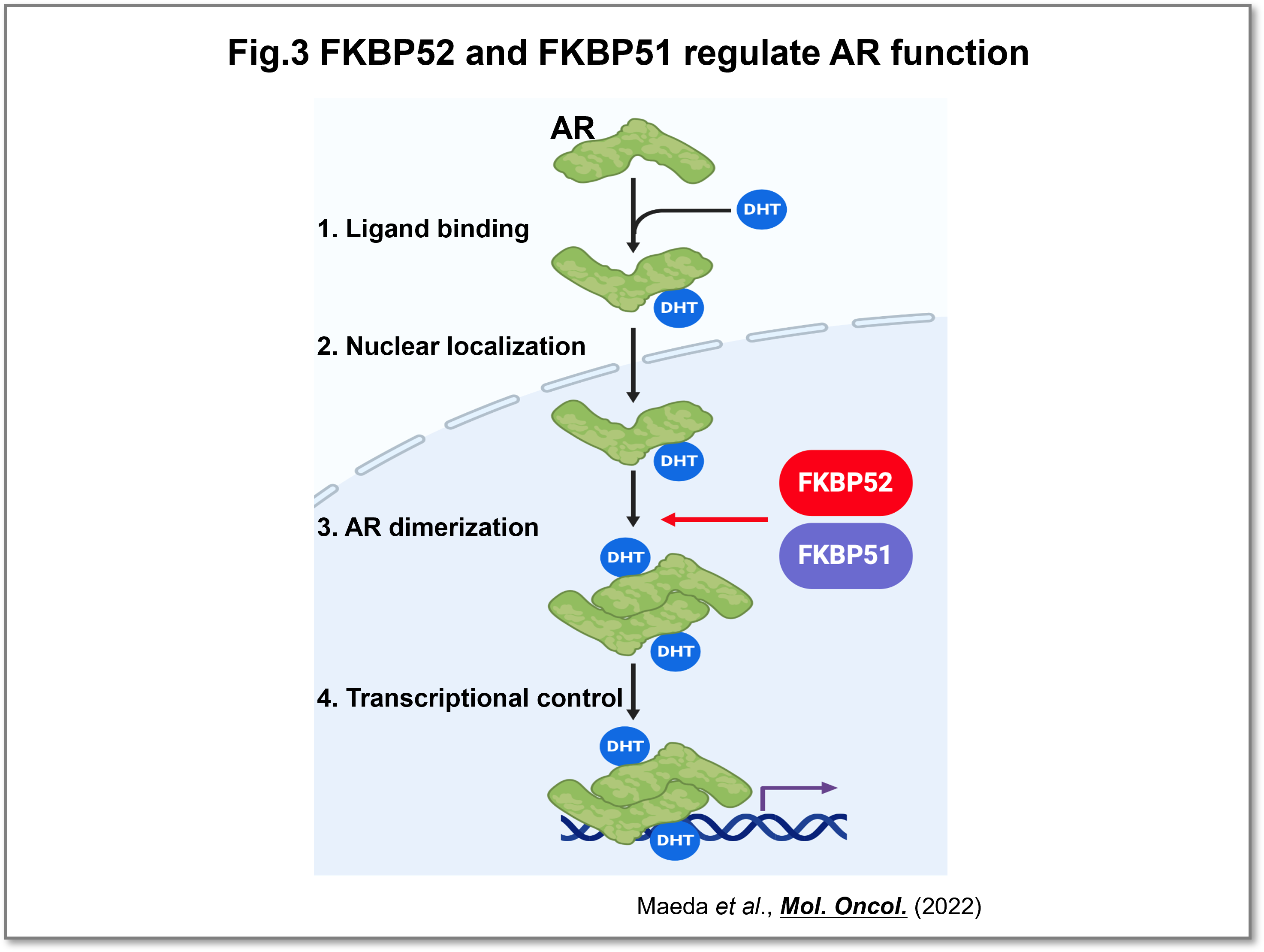
(画像をクリックで拡大)
プロリン異性化がもたらす生命現象の理解
プロリン異性化酵素は複製、転写、翻訳、活性、局在、分解と言った様々な生理機能に関与している可能性が示されています(Fig.4)。またその異常はがん、老化、炎症、不妊、神経変性疾患などの病態と関連していることがこのように多数知られており、新たな研究が進みつつあります。しかしプロリンの異性化が、実際にどの程度立体構造に影響を与えるのか、どのような機序で生理機能に影響を与えるのかを示した研究は限られています。今までの生物学では、リン酸化、ユビキチン化などを中心としたタンパク質の化学修飾が注目され研究されてきましたが、それらの化学修飾に加えて、プロリン異性化による立体構造変化が複雑な生命現象を司っているのではないかと考えています。このことに関してはまだ重要性、意義、メカニズムについてはわからないことが多く、現在研究を進めています。
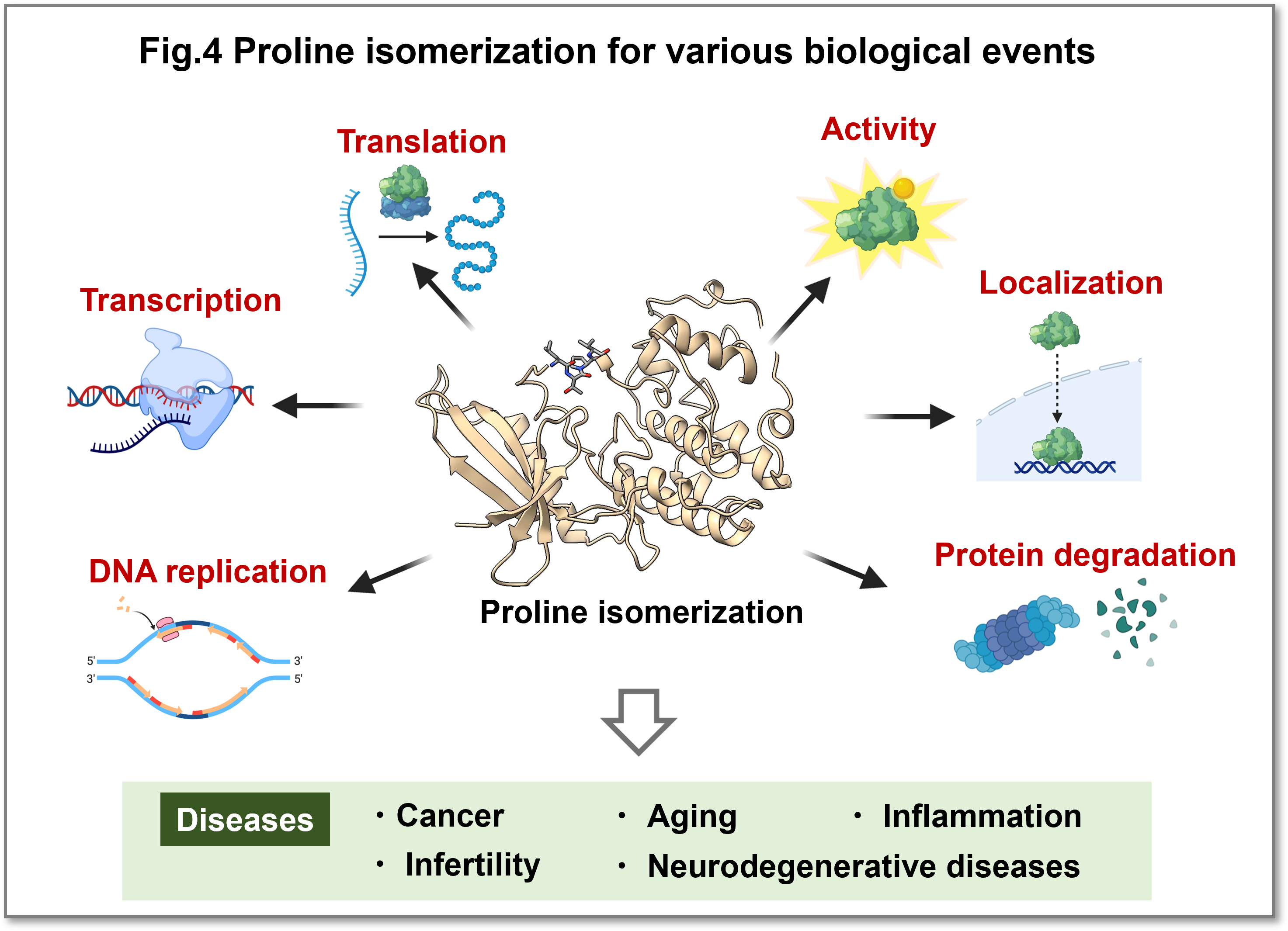
(画像をクリックで拡大)
カルシウムシグナルの破綻がもたらす細胞増殖異常とがん
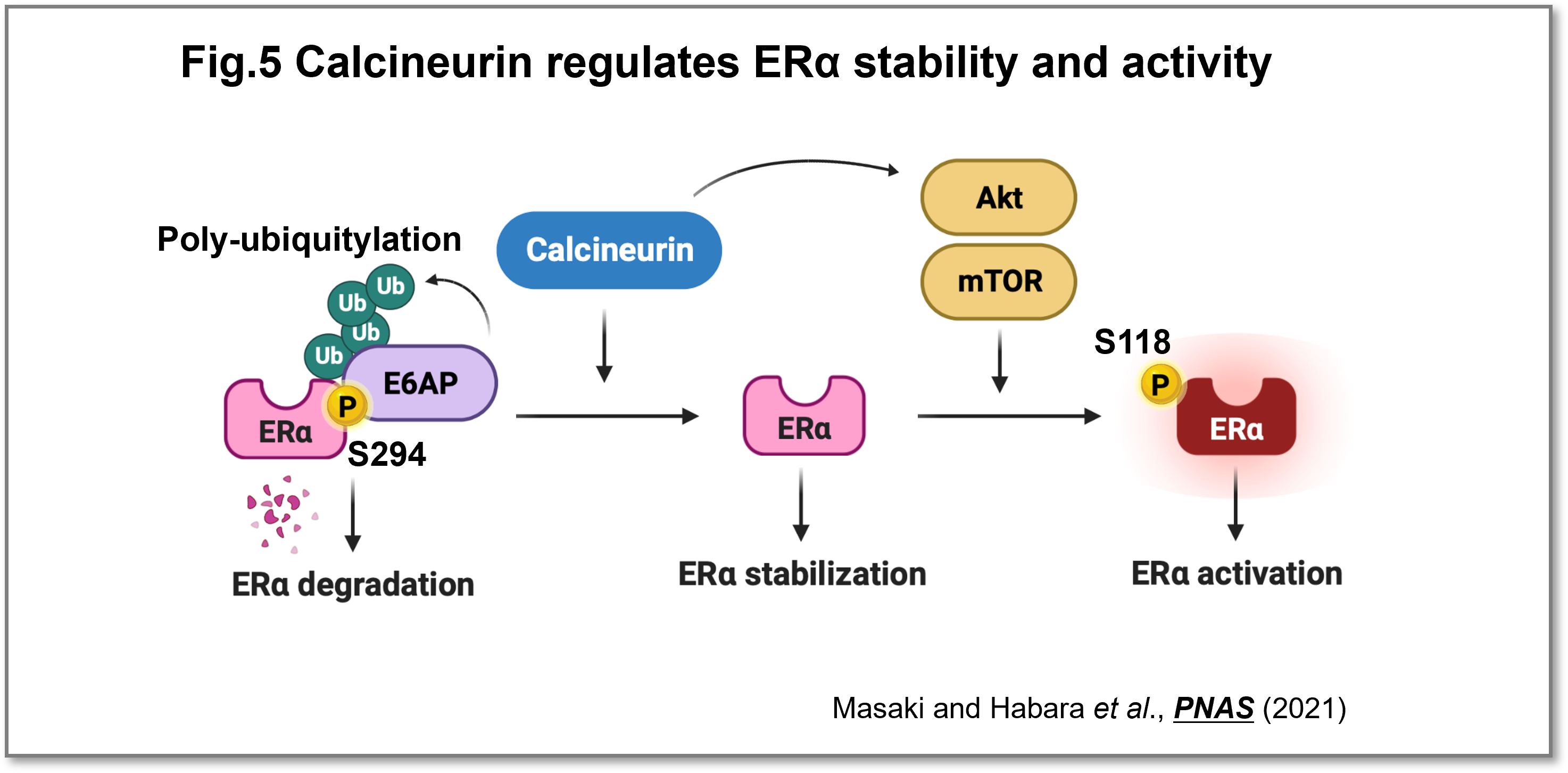
乳がんの再発に関わる新たな制御因子としてカルシニューリンを発見し、悪性腫瘍の再発に密接に関連するエストロゲン受容体の機能をカルシニューリンが増強するメカニズムを明らかにしました(Masaki and Habara et al.,PNAS, 2021)(Fig.5)。カルシニューリンはカルシウムシグナルを仲介する重要な脱リン酸化酵素であり、免疫抑制剤FK506の標的分子として臨床的にも重要であることが知られています。カルシニューリンはがん細胞の増殖を促進させる機能を持つことが報告されていましたが、がん患者さんの予後との相関関係については未解明な状況にありました。本研究では、再発性乳がんに着目し、カルシニューリンが高発現していると内分泌治療後の乳がんの再発率が高くなり、予後不良となることを発見しました。カルシニューリンは、(1)エストロゲン受容体を脱リン酸化することで、エストロゲン受容体の分解を防ぐこと、(2)mTORキナーゼを介してエストロゲン受容体の活性を促進すること、の2つの作用により、エストロゲン受容体の機能を増強することが判明しました(Fig.5)。今回の研究成果は、エストロゲン受容体陽性乳がんの患者さんに対して抗エストロゲン療法を施した場合、再発するメカニズムに対して理解を深めることとなり、新たなバイオマーカー、効果的な治療法開発へと展開することが期待できます。
(画像をクリックで拡大)
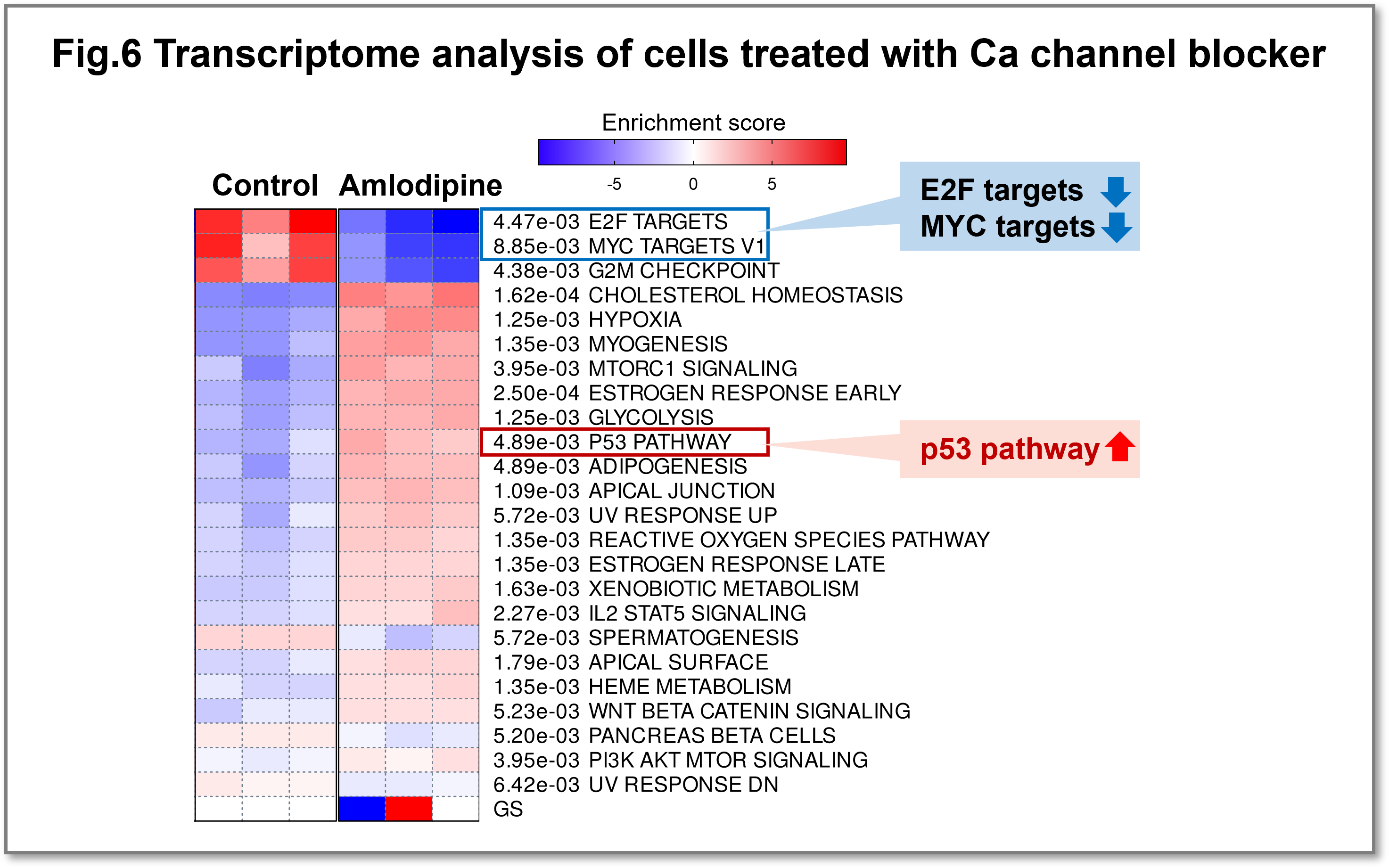
細胞内カルシウムイオン(Ca2+)は、シグナル伝達経路において多面的なセカンドメッセンジャーとして作用し、遺伝子発現、細胞周期、細胞運動、オートファジー、アポトーシスなど様々な生命現象に関与しています。Ca2+の役割は多岐にわたるため、カルシウム恒常性の異常は様々な疾患と関連する。がん細胞では、細胞内のCa2+濃度の変化が腫瘍の発生、血管新生、進行、転移に関与していると考えられていますが詳細は不明です。私達はCa2+ががん細胞の増殖に与える影響を明らかにするために、まずCa2+チャネル阻害薬Amlodipine投与時における網羅的な遺伝子発現変化を検討しました。その結果、Amlodipine投与により細胞内Ca2+濃度が低下すると、(1)がんの悪性化に寄与するc-Myc、E2F標的遺伝子の発現が減少すること、(2)p53標的遺伝子の発現が増加することがわかりました(Fig.6)。
(画像をクリックで拡大)
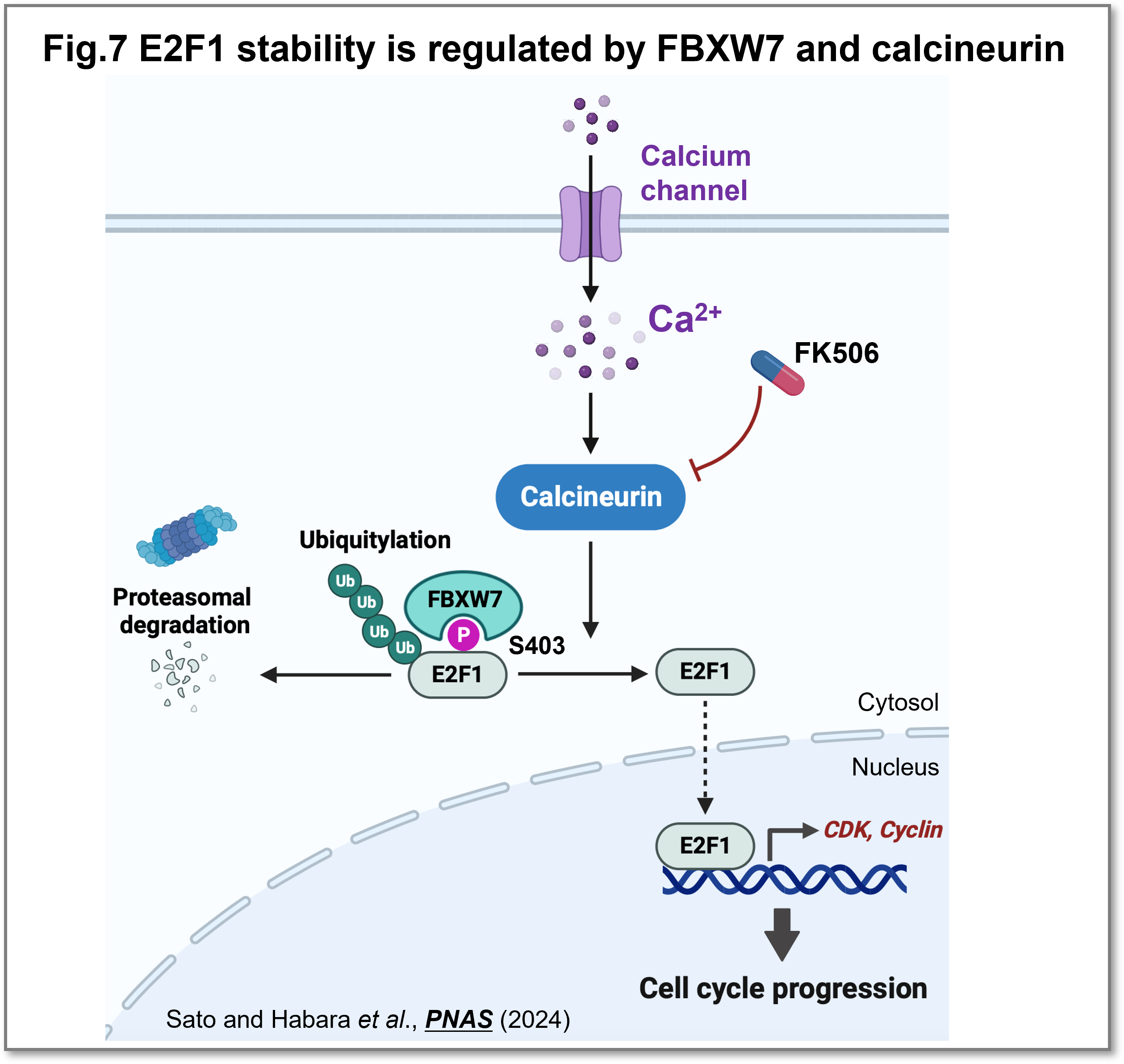
E2F1は細胞周期を促進し、がん細胞での異常な増殖に寄与するため、その分解を制御することはがん治療の重要な戦略となり得ます。私達は、ユビキチン化酵素複合体のサブユニットであるFBXW7がE2F1をユビキチン化し、分解を促進することを発見しました(Sato and Habara et al.,PNAS, 2024)(プレスリリース)。細胞内カルシウム量が増加するとCalcineurinが活性化されE2F1を脱リン酸化することで、FBXW7との結合を抑制し、E2F1を安定化することを明らかにしました(Fig.7)。本研究によって、カルシウムチャネルやカルシニューリンの阻害が新たながん治療戦略となりうる可能性が期待されます。細胞内カルシウム量によってCalcineurinがc-Mycを脱リン酸化し安定化すること、NFATcの核内移行を促進することでMDM2の転写を活性化することも明らかにしました(Masaki et al.,Sci. Rep., 2023) (Hanaki et al.,Life Sci. Alliance, 2024)。
(画像をクリックで拡大)
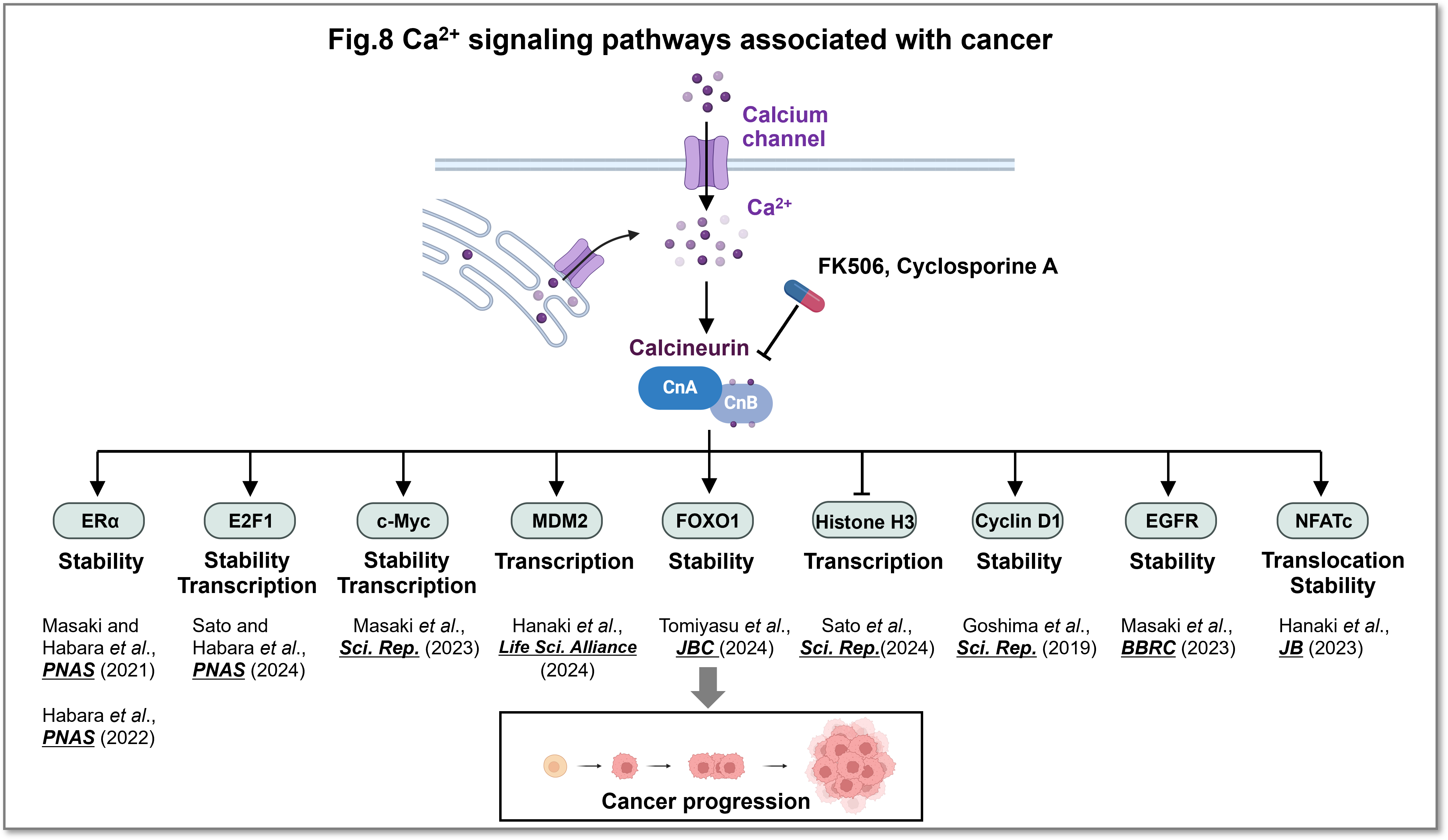
カルシウムシグナルの異常は発がんと密接に関連していますが、まだ理解は十分ではありません。私達は、カルシウムシグナルの変化ががんの発生、悪性化にどのように影響するのかを明らかにしたいと考えて研究しており、がんの悪性化に関わるカルシニューリンの標的を複数見出しています(Fig.8)。今後、さらなる解析を行い、カルシウムシグナルを標的としたがん治療法の確立へと展開させていきたいと考えています。
(画像をクリックで拡大)
ゲノム安定性維持機構解明
ヒストンH3-T11のリン酸化による増殖関連遺伝子の転写制御
Chk1の変異は、ゲノム不安定化を引き起こして発がんを誘発します。一方でChk1は特定のがんでは高活性化しており、細胞増殖促進やがんの悪性度に寄与します。これまで不明であったChk1の細胞増殖を促進する機能として、ヒストンのリン酸化を介した増殖関連遺伝子の転写活性化を発見しました(Shimada et al.,Cell, 2008) (Shimada et al.,Cell Cycle, 2008)。またPP1脱リン酸化酵素がDNA損傷後に活性化されることで、増殖関連遺伝子の転写を抑制することを見出しました(Shimada et al.,EMBO Rep., 2010)。さらにPP1の活性調節に重要な制御因子としてNIPP1を同定し、その調節機構を明らかにしました(Hanaki et al.,Cancer Sci., 2021)(Fig.9)。
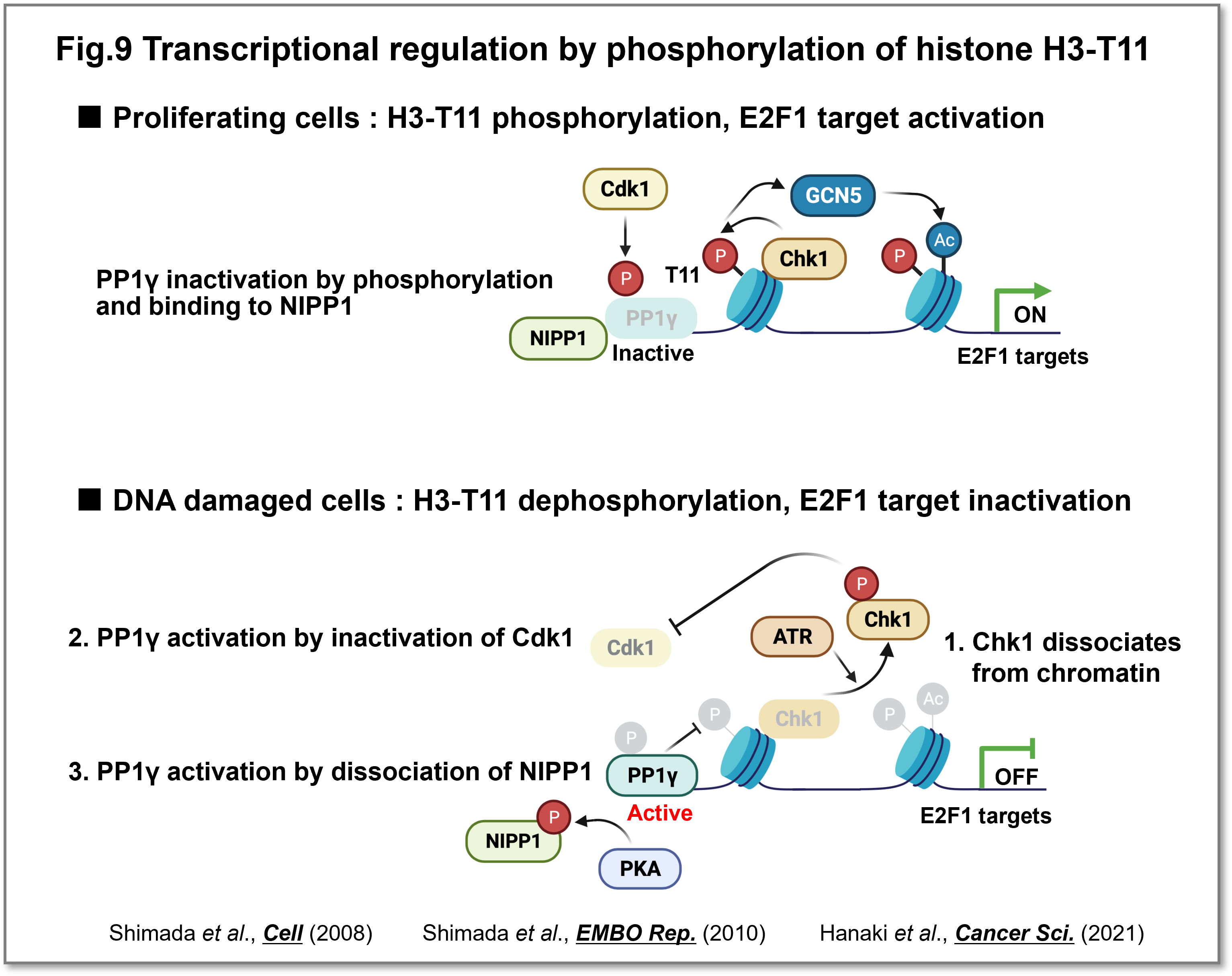
(画像をクリックで拡大)
ヒストンH2AX-S121のリン酸化を介した染色体分配制御
細胞分裂期における娘細胞への均等な染色体分配は、安定的な染色体維持に必須であり、AuroraBキナーゼが染色体分配の制御に中心的な役割を果たしています。細胞周期の進行に伴いAuroraBの活性および局在が厳密に制御されることが重要ですが、どのようにAurora Bの活性化がセントロメア局所的かつ急激に起こるのかについては不明でした。私達はDNA損傷応答に重要なヒストンH2AバリアントのH2AXが、DNA損傷応答の機能とは無関係に染色体分配に必要であることを明らかにしました。さらにH2AX-S121は活性化型AuroraBのセントロメアへの効率よいリクルートに関わり、AuroraBの時空間的な活性化に重要であることを示しました(Shimada et al.,Nat. Commun., 2016)(Fig.10)。
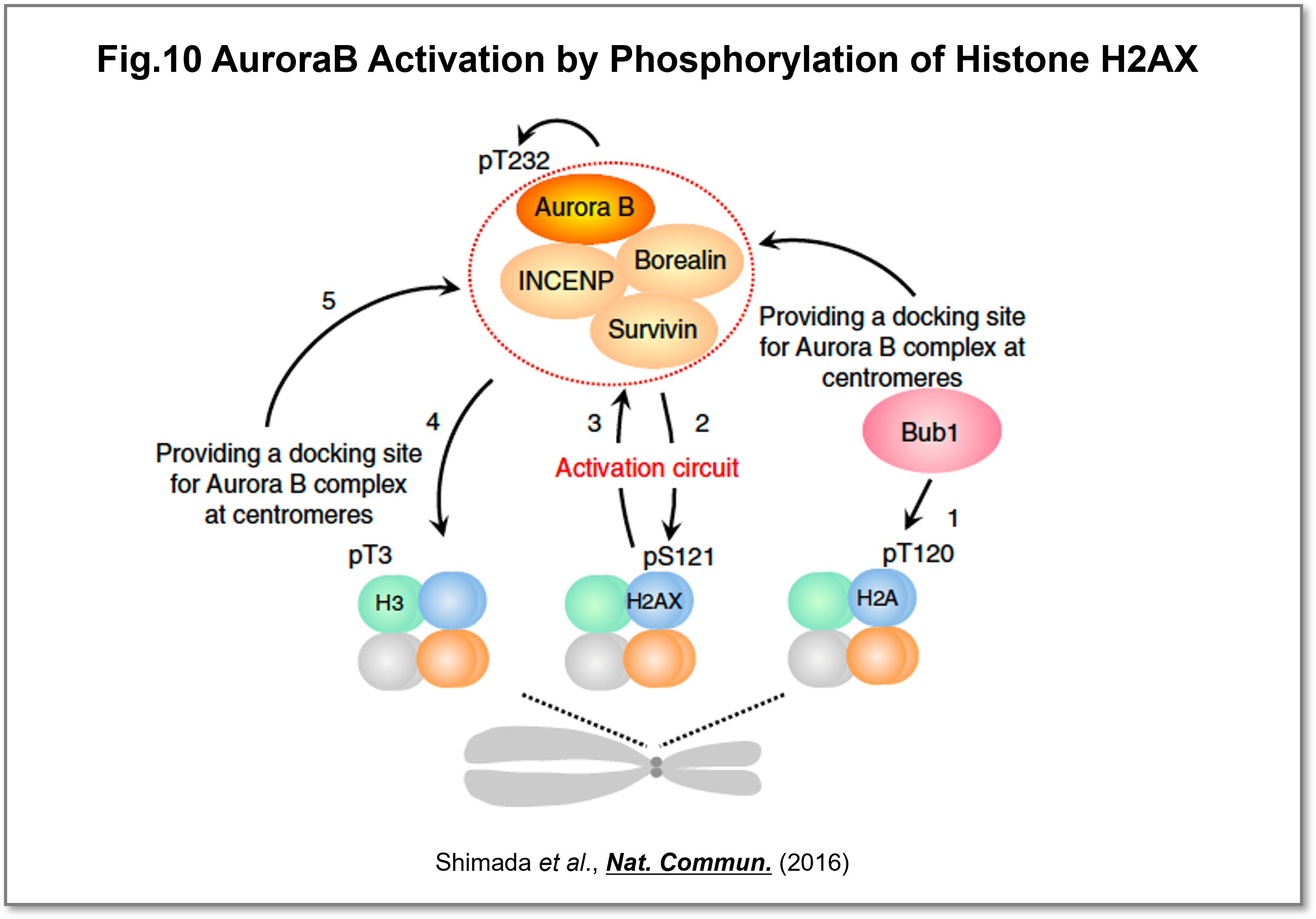
(画像をクリックで拡大)
減数分裂組換えチェックポイント機構の発見
減数分裂の中で相同染色体の組換えは、遺伝的多様性を生み出す以外にも組換え過程で生じるキアズマ構造が正常な減数分裂期の染色体分配に必須です。細胞周期チェックポイントは体細胞分裂では詳しく調べられていましたが、減数分裂ではほとんど分かっていませんでした。私達は分裂酵母の減数分裂期組換え時に生じるDNA二本鎖切断が修復されるまで減数第一分裂を抑制するチェックポイント機構が存在し、正常な配偶子形成を保障することを明らかにしました(Shimada et al.,EMBO J., 2002)(Fig.11)。
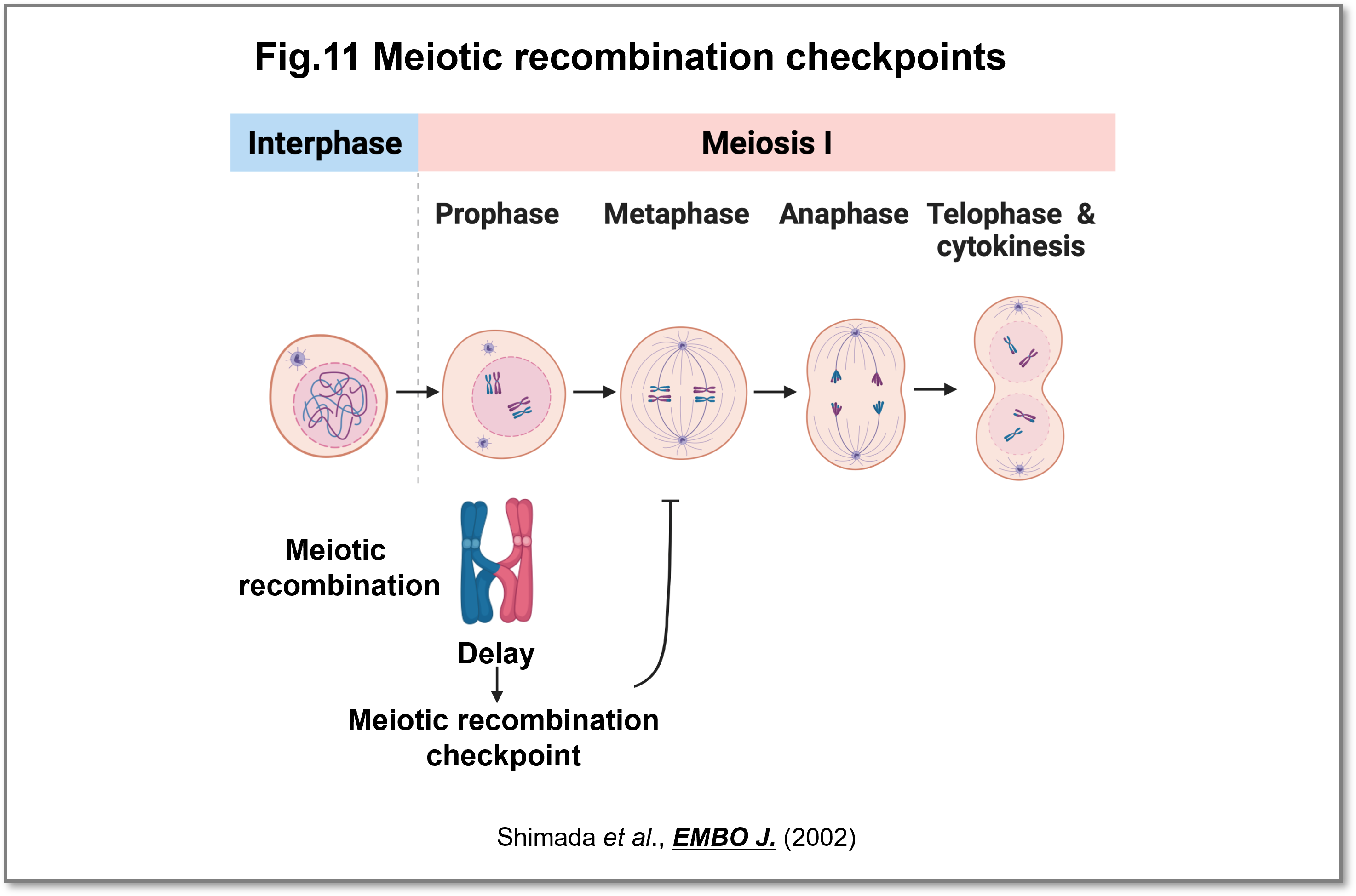
(画像をクリックで拡大)
体細胞分裂から減数分裂への移行の分子機構の解明
体細胞分裂の細胞周期はCdkにより制御され、その分子機構は酵母からヒトまで保存されていますが、体細胞分裂から減数分裂への移行の分子機構はほとんど理解されていませんでした。分裂酵母においてフォークヘッド型転写因子であるFkh2は、Cdk1によってリン酸化されると、減数分裂への進行に必要なste11の転写が阻害されることを示しました。体細胞分裂から減数分裂への移行には、Cdk1が不活性化されることでFkh2のリン酸化が減少し、stellの転写を活性化することを明らにしました。本研究は、体細胞分裂から減数分裂への移行の制御機構の理解に貢献しました(Shimada et al.,EMBO J., 2008)(Fig.12)。
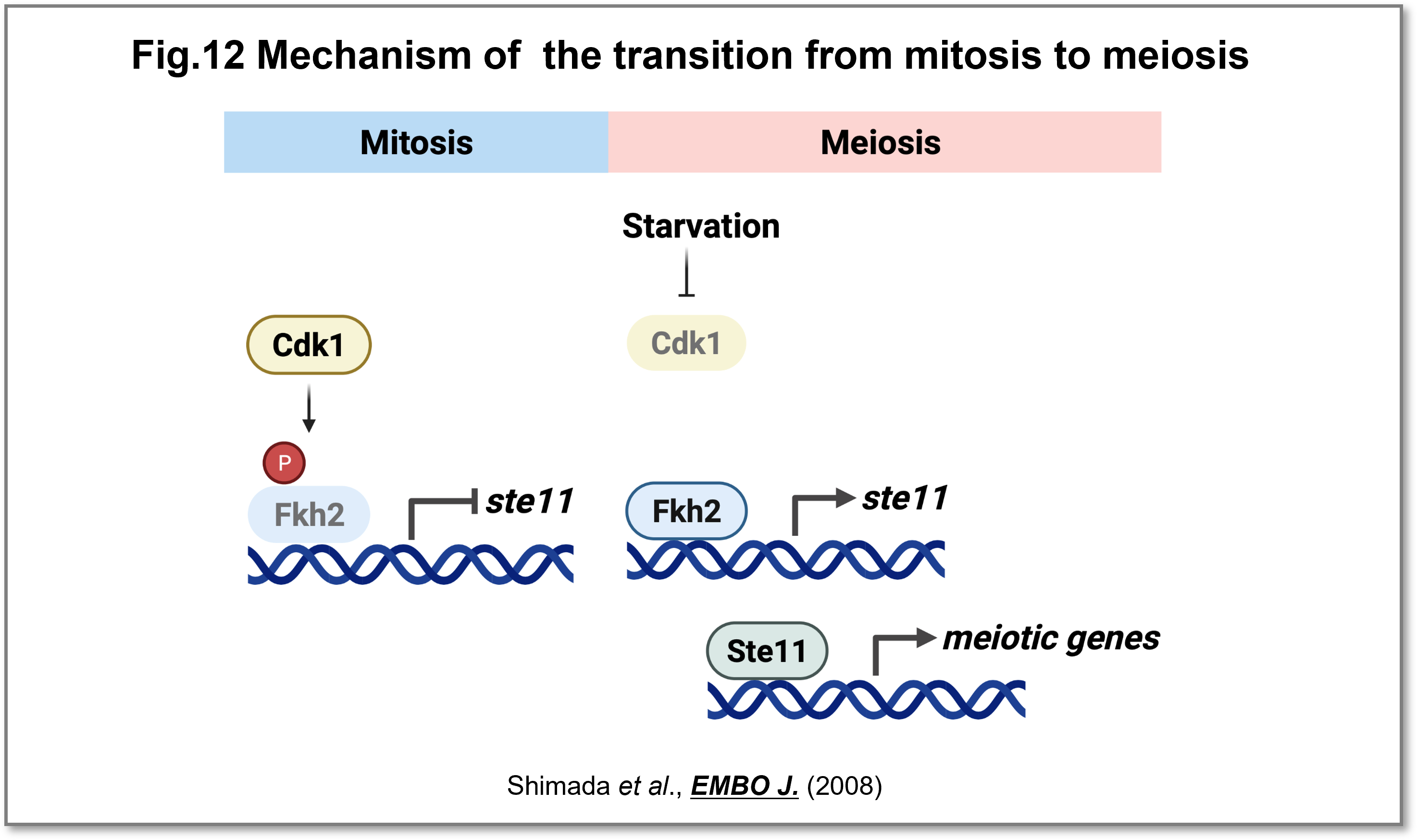
(画像をクリックで拡大)