研究室紹介Laboratories
- Back
- Top > 研究室紹介 > 先端応用医学(協力) > 神経遺伝情報学
先端応用医学(協力)神経遺伝情報学
研究室概要
当分野は、2004年9月に神経疾患腫瘍分子医学センターの一部門としてスタートしました。(1) 神経筋接合部ならびに筋収縮に伴う信号伝達系の正常分子機構・分子病態・新規治療法開発研究、(2) RNAスプライシングを中心としたRNA代謝の正常分子機構と病態解明研究、(3) ドラッグリポジショニング戦略による神経・筋・骨格疾患の治療法開発、(4) 腸内細菌叢解析によるパーキンソン病の病態機構解明、(5) 分子状水素の酸化ストレス病態を中心とする多彩な疾患に対する効果とその分子作用機構解明の研究を行っています。
研究プロジェクト
1. 神経筋接合部ならびに筋収縮に伴う信号伝達系の正常分子機構・分子病態・新規治療法開発研究
i) 先天性筋無力症候群(congenital myasthenic syndromes, CMS)
CMSは、神経筋接合部情報伝達の障害により病的な筋力低下と易疲労性が生じる疾患群である(図1) (eLS DOI: 10.1002/9780470015902.a0024314, 2014)。CMSにおいて24種類の遺伝子(図1に赤字で示す分子群)に変異が報告されている。24種類の遺伝子は、骨格筋アセチルコリン受容体(AChR)サブユニットをコードするCHRNA1, CHRNB1, CHRND, CHRNE CHRNG; 骨格筋ナトリウムチャンネルをコードするSCN4A; AChRのクラスタリングを誘導する分子群AGRN, LRP4, MUSK, DOK7; 神経筋接合部シナプスの構造分子COLQ, LAMB2 COL13A1; 筋終板の構造分子RAPSN, PLEC; シナプス前分子CHAT, SYT2; グリコシレーション酵素群GFPT1, DPAGT1, ALG2, ALG14, GMPPB; 機能不明の分子群PREPL, SCL25A1である。CMSはAChRサブユニットミスセンス変異によるスローチャンネル症候群とsynaptotagmin 2遺伝子(SYT2)変異による先天性Lambert Eaton筋無力症候群を除いて常染色体劣性遺伝形式である。
CMSの多くは2歳以下で発症するが成人発症も稀ではない。特に優性遺伝形式を取るスローチャンネル症候群と、グリコシレーション酵素欠損ならびにDOK7変異による肢体型CMSでは成人発症例が多い。CMSの臨床症状は、日内変動・日差変動を伴う筋易疲労性、骨格筋低形成、軽度顔面奇形を特徴とする。外眼筋、顔面筋、球筋が侵されることが多い。我々は、(1) 神経終末に取り込まれたコリンからアセチルコリンを再合成するcholine acetyltransferase (ChAT) (Proc Natl Acad Sci U S A 98: 2017, 2001)、(2) acetylcholinesteraseをシナプス基底膜にanchoringをするcollagen Q (Proc Natl Acad Sci U S A 95: 9654, 1998)、(3) リガンド結合性イオンチャンネルのアセチルコリンレセプター(AChR) (Proc Natl Acad Sci U S A 92: 758, 1995, Neuron 17: 157, 1996)、(4) AChRを終板にclusteringさせるrapsyn (Am J Hum Genet 70: 875, 2002)、(5) AChRによる終板電位を感知し筋活動電位を起こす骨格筋電位依存性ナトリウムチャンネルNaV1.4 (Proc Natl Acad Sci U S A 100: 7377, 2003)、(5) 筋終板におけるAChR集積誘導シグナルを伝えるLRP4 (Hum Mol Genet 23: 1856, 2014, JAMA Neurol 72: 889, 2015)の遺伝子変異を世界に先駆けて同定しその機能解析を行ってきた。
2009年に我々が本邦の遺伝子診断を開始する前には本邦で遺伝子変異を同定できた症例は1例のみであった。主に次世代シークエンサによるエキソーム解析により本邦約20症例の遺伝子診断を行ってきた。我々はCOLQ遺伝子変異がColQのMuSKへの結合を阻害することを明らかにした(図2, 3) (Hum Mutat 34: 997, 2013)。加えて、変異ColQを用いたColqノックアウトマウスに対するタンパク係留治療法(後述)が無効であることを実証した(図4)。さらに、スローチャンネル症候群と終板AChR欠損症の症例群を報告した(図5) (Neuromuscul Disord 25: 60, 2015)。AChRεサブユニットの変異によるスローチャンネル症候群では単一イオンチャンネル解析にてAChRのイオンチャンネル孔を構成するvaline ringがAChRイオンチャンネル動態に重要であることを明らかにした(図6) (Hum Mutat 37: 1051, 2016)。我々はさらに新しい分子の遺伝子変異を調べるとともにその分子作用機構の解明を精力的に行っている。
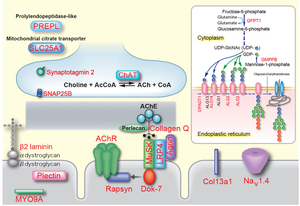 図1. 先天性筋無力症候群において欠損する24種類の神経筋接合部分子。 |
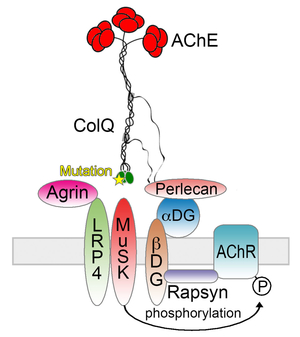 図2. ColQはMuSKとperlecanに結合してシナプス基底膜に係留する。 |
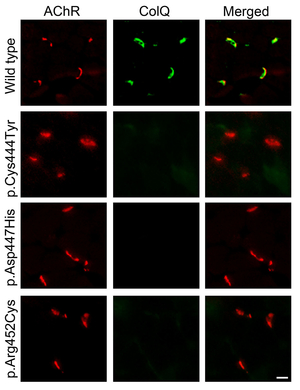 図3. 変異ColQはColqノックアウトマウス神経筋接合部に係留できない。 |
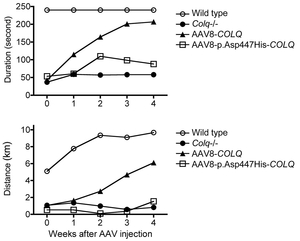 図4. AAV8によるタンパク係留療法をColqノックアウトマウスに対して行ったところp.D447H変異ヒトCOLQは正常ヒトCOLQと異なりマウス運動障害を改善できなかった。 |
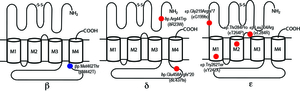 図5. 本邦の先天性筋無力症候群において同定されたアセチルコリン受容体の遺伝子変異(赤丸)。単一イオンチャンネル解析によりβサブユニット変異(青丸)は日本人特有の正常ポリモリフィズムであった。 |
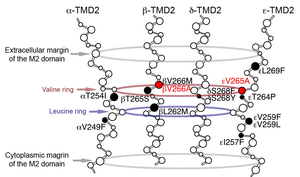 図6. アセチルコリン受容体イオンチャンネル孔のバリンからアラニンへの変異はスローチャンネル症候群を起こす。 |
ii) タンパク係留治療法
遺伝子治療はモデル細胞においてはあらゆる病態の治療が実質的に可能である。しかし、遺伝子の標的臓器特異的導入が困難なため遺伝子治療のヒトへの応用は限られている。細胞外マトリックス分子(ECM)は標的臓器に係留するための固有のドメインを有している。我々はこのECMの特徴を生かして先天的なECM欠損病態に対するECM補充を行いタンパク係留療法と名付けた。
終板アセチルコリンエステラーゼ(AChE)欠損症はCOLQ遺伝子変異を原因とする。本病態に対する治療法は存在しない。非対称性AChE/ColQ複合体(図7)はECMであり、ColQのコラーゲンドメインとC末端ドメインによりシナプス基底膜に係留する(図8)。AAV8-COLQはColqノックアウトマウスに対して顕著な運動能改善・神経筋接合部信号伝達改善効果を認めた(図9, 10, 11) (Mol Ther 20: 1384, 2012)。さらに、デュシャンヌ型筋ジストロフィーモデルmdxマウスに対して、ECM分子biglycanのAAV8による係留を行い筋病理所見ならびに運動障害が改善することを明らかにした(図12) (Hum Gene Ther in press, 2016)。
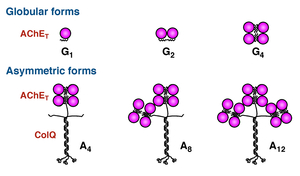 図7. 骨格筋の6種類のアセチルコリンエステラーゼ分子。 |
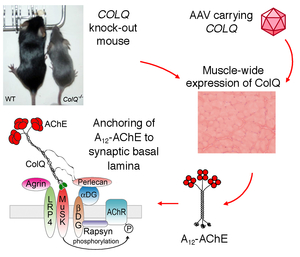 図8. Colqノックアウトマウスに対するタンパク係留療法。 |
 図9. タンパク係留療法によりColqノックアウトマウスが活発になった[movie]。 |
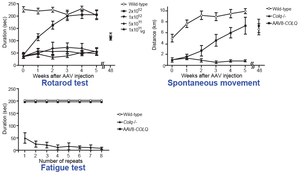 図10. タンパク係留療法によるColqノックアウトマウスの運動機能改善効果。 |
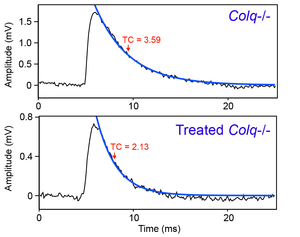 図11. タンパク係留療法により微小終板電位(MEPP)減衰時間が短縮した。 |
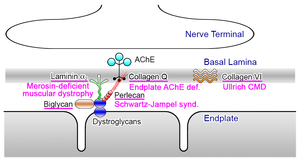 図12. タンパク係留療法が有効であることが想定される細胞外マトリックス分子。 |
iii) アセチルコリン受容体(AChR)クラスタリングを誘導する新規分子の同定
神経筋接合部においてAChRクラスタリングを誘導する新規分子を同定する目的でマウス脊髄のレーザーキャプチャマイクロダイセクションを行いマイクロアレイならびにRNA-seqにより解析を行った(図13)。分泌型Wnt活性化因子R-spondin 2 (Rspo2)が脊髄前角細胞(SMN)に特異的に発現し、神経筋接合部に係留していることを見いだした。培養細胞においてRspo2はagrin非存在下でMuSKリン酸化を促進し、AChRクラスタリングを誘導した(図14)。Lgr5 (leucine-rich repeat-containing G-protein coupled receptor 5)がMuSKとLRP4に結合し神経筋接合部に高く発現することを見いだすとともにRspo2の受容体であることを明らかにした(図15) (Sci Rep 6: 28512, 2016)。我々はさらに2つの新規AChRクラスタリング誘導分子の研究を続けている。
 図13. マウス脊髄前角細胞のレーザーキャプチャーマイクロダイセクションによりRspo2が脊髄前角細胞に高発現であることを明らかにした。 |
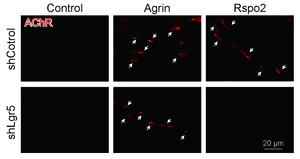 図14. Rspo2はagrin非存在下にアセチルコリン受容体のクラスタリングを誘導する。Lgr5受容体のノックダウンによりRspo2の効果は消失する。 |
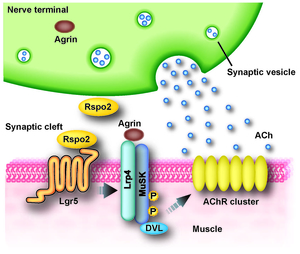 図15. Rspo2によるアセチルコリン受容体クラスタリング誘導の概念図。 |
iv) 重症筋無力症における抗MuSK抗体の標的分子の同定
重症筋無力症は抗AChR抗体、抗MuSK抗体、抗LRP4抗体が原因である。生理的な条件においてMuSKはLRP4とColQに結合する。上述のようにColQはアセチルコリンエステラーゼ(AChE)と複合体を形成し、AChEをシナプス基底膜に係留する。我々は抗MuSK抗体がColQのMuSKへの結合を阻害することを見いだし、患者由来抗MuSK抗体の正常マウスへの移入によりAChE/ColQ複合体が減少することを報告した(図16) (Neurology 77: 1819, 2011)。さらに、抗MuSK抗体は、agrin存在下においてLRP4とMuSKの結合も阻害することを見いだし、患者由来抗MuSK抗体のColqノックアウトマウスへの移入によりAChR欠損が誘導されることを報告し、AChR欠損には抗MuSK抗体によるColQとMuSKの結合阻害は関与しないことを明らかにした(Sci Rep 5: 13928, 2015)。さらに、驚くべきことに、正常においてColQはMuSKに結合することにより agrin/LRP4/MuSK信号伝達系を阻害することが判明した。定量的な解析にて抗MuSK抗体はColQよりも高度にagrin/LRP4/MuSK信号運伝達系を阻害することによりAChR欠損を起こしていた(図17)。我々はagrin, LRP4, MuSK, ColQ, biglycanと抗MuSK抗体の相互関係をドメイン構造を含めてさらに詳細に調べている。
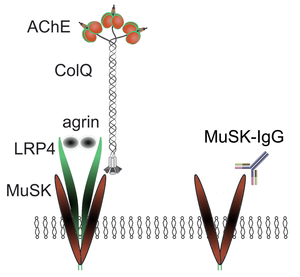 図16. 抗MuSK抗体はColQのMuSKへの結合を阻害する。 |
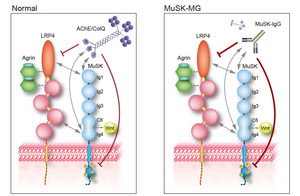 図17. アセチルコリンエステラーゼ(AChE)/ColQ複合体はMuSKとColQの結合を阻害しMuSKリン酸化を阻害する。抗MuSK抗体はAChE/ColQ複合体をMuSKから退けるとともにAChE/ColQ複合体よりも強くMuSKリン酸化を阻害する。 |
2. RNAスプライシングを含むRNA代謝の正常分子機構と病態解明研究
i) スプライシングシス因子の解明とシス因子を破断する遺伝子変異予測ツールの開発
ヒトは約18,000個という限られた数の遺伝子から多様なタンパク質を作るために、組織特異的・発達段階特異的なalternative splicingを行っている。96%以上のヒトのマルチエクソン遺伝子がalternative splicingを受ける。Alternative splicingはexon上を含む各遺伝子上のsplicing cis-elementsと、組織特異的・発達段階特異的に発現するsplicing trans-factorsによりコントロールされている(図18)。
クラシカルなsplicing cis-elementsには、branch point sequence, polypyrimidine tract, 3’ splice site, 5’ splice siteが知られている。これらのcis-elementsはdegenerativeなため、これらを破壊する遺伝子変異のsplicingに与える影響の予測は困難であった。我々は5’ splice siteのsplicing signalの強さを評価するSD Scoreを開発しweb service programを提供している(図19) (Nucleic Acids Res 35: 5995, 2007)。また、ヒトのbranch point consensus sequnceは、酵母で保存されたbranch point配列UACUAACとの類似性から導き出されていたのみで、branch point sequenceを破断する遺伝子変異のsplicingに与える効果は予測困難であった。我々は多数のlariat RT-PCR産物の遺伝子配列解析によりヒトbranch point consensus sequenceがyUnAyであることを明らかにした(図20) (Nucleic Acids Res 36: 2257, 2008)。さらに、ポリピリミジンが短いAG-dependent 3’ splice sitesにおいてはエクソン第1塩基の塩基置換によりU2AF35の結合が阻害をされスプライシング異常を起こすことを明らかにした(図21) (Nucleic Acids Res 39: 4396, 2011)。さらに、intron 3’末端polypyrimidine tractを含むintron -50位から-3位の塩基置換がsplicingに影響を与えるかを評価するツールIntSpliceを開発しweb service programを提供している(図22) (J Hum genet 2016)。塩基置換のスプライシングへの影響を解析ツールの開発を行っている研究者は世界的にも極めて少なく我々のスプライシング予測ツール開発研究は、次世代シークエンサによって大量に生み出される塩基置換の病的意義を検討するツール群として役立っている。我々は同様の解析手法により、偽エクソンの活性化予測や、アミノ酸置換の病的意義を検討するツールの開発を現在行っている。
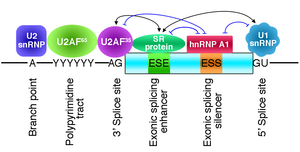 図18. スプライシングシス因子とRNA結合タンパク。 |
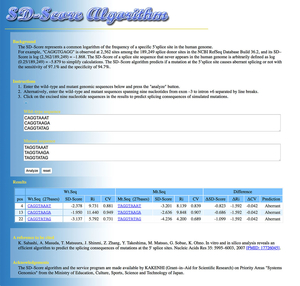 図19. SD-Scoreは5’スプライスサイトの塩基置換のスプライシングに与える影響を感度97.1%、特異度94.7%で予測する。 |
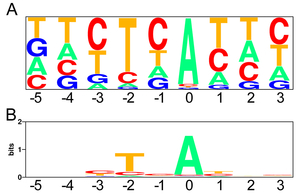 図20. ヒトのブランチポイント配列のピクトグラム(A)とWebLogo (B)。 |
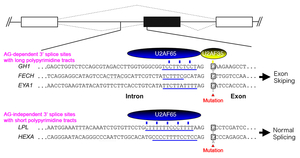 図21. ポリピリミジントラクトが短いAG依存性3’スプライスサイト(AG-dependent 3’ splice site)を持つエクソンの第1塩基の塩基置換はスプライシング異常を起こす。 |
 図22. IntSpliceウェブサービスプログラムはイントロン3’末端領域-50位から-3位の塩基置換のスプラシングに与える影響を予測する。 |
ii) Splicing cis-elementsを破断する遺伝子変異の分子病態機構解明
我々はsplicing cis-elementsを破断する遺伝子変異の解析を行ってきた。アセチルコリン受容体αサブユニットをコードするCHRNA1遺伝子のイントロン領域の遺伝子変異がRNA結合タンパクhnRNP Hの結合を減弱しスプライシング異常を起こすことを明らかにした(図23) (Hum Mol Genet 17: 4022, 2008)。さらに、同部位にはポリピリミジントラクト結合タンパク(PTB)も結合することを明らかにした(Hum Mol Genet 18: 1229, 2009)。別の患者においてCHRNA1のエクソン上の点変異がhnRNP Lの結合を減弱し、hnRNP LLの結合能を新たに得ることによってスプライシング異常を起こすことを明らかにした(図24) (Sci Rep 3: 2931, 2013)。同様にcollagen QをコードするCOLQ遺伝子のエクソン16の点変異がSRSF1の結合を減弱するとともに新たにhnRNP Hの結合能を得ることによりエクソンスキッピングを起こすことを報告した(図25) (Sci Rep 5: 13208, 2015)。我々は先天性筋無力症候群ならびに他の疾患におけるsplicing cis-elementsを破断する遺伝子変異の分子病態機構研究をさらに続けている。
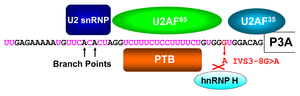 図23. CHRNA1エクソンP3AのスプライシングはhnRNP HとPTBによって抑制される。先天性筋無力症候群におけるG-to-A遺伝子変異はhnRNP Hの結合を減弱しエクソンP3Aが常に含まれる。 |
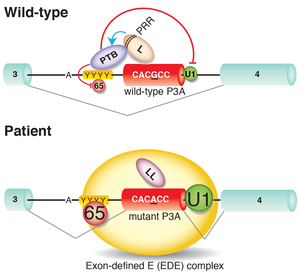 図24. hnRNP L (L)のproline-rich region (PRR)はPTBに結合しCHRNA1エクソンP3Aのスプライシングを抑制する。先天性筋無力症候群におけるG-to-A遺伝子変異はhnRNP Lの結合を減弱しhnRNP LL (LL)の結合能を獲得することによりエクソンP3Aが常に含まれる。 |
 図25. COLQエクソン16は選択的なスプライシングを受けないにもかかわらずSRSF1によりスプライシングが増強される。先天性筋無力症候群における遺伝子変異はSRSF1の結合を阻害しhnRNP Hの結合能を獲得することによりエクソンスキッピングを起こす。 |
iii) 神経筋接合部に発現する遺伝子の選択的プライシングの分子機構解明
MUSK遺伝子exon 10はヒトにおいて選択的スプライシングを受けWntによるアセチルコリン受容体のクラスタリングに重要なシステインリッチ領域(Fz-CRD)を欠損したMuSKが作られる(図26)。興味深いことにマウスMuskにはこの選択的スプライシングはない。ヒトMUSK exon 10にhnRNP C, YB-1, hnRNP Lが結合しエクソンスキッピングを惹起することを見いだした(Sci Rep 4: 6841, 2014)。hnRNP CがYB-1とhnRNP LのRNA結合を促進し、3つのRNA結合タンパクが相加的に作用をする。加えて、ゲノムワイドの検索により類似の制御を受けるエクソンを複数見いだしている。
ACHE遺伝子によりコードされるアセチルコリンエステラーゼ(AChE)は神経伝達物質アセチルコリンを分解し神経筋接合部の信号伝達を止める役割を持つ。ACHE遺伝子の3’末端近傍の選択的スプライシングによりAChE-T, AChE-H, AChE-Rの3種類のアイソフォームが作られる(図27)。我々はhnRNP Hがexon 5aの2カ所のG runsに結合し、exon 5aとexon 5bの境界の遠位部の3’スプライスサイトを活性化することによりAChE-Tアイソフォームが作られることを明らかにした(図28)。さらに、hnRNP HはCstF64とexon 5aへの結合を競合しexon 5a上の選択的なポリAサイトの活性化を抑制することを見いだした。加えて3種類の細胞株においてhnRNP Hの発現レベルがAChE-Tアイソフォームの発現量と正相関していた。さらに、ゲノムワイドの解析にて複数の遺伝子においてhnRNP Hが選択的3’スプライスサイト活性化と選択的ポリAサイト活性化の競合を制御していることを見いだした(Nucleic Acids Res in press)。我々は神経筋接合部に発現する他の遺伝子においてもスプラシング調整の正常分子機構の解明研究を精力的に行っている。
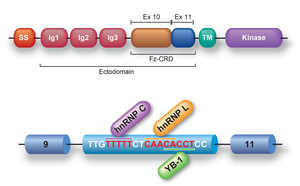 図26. MuSKのFz-CRDドメインはエクソン10と11によりコードされる。MUSKエクソン10へのhnRNP Cの結合は、その下流へのhnRNP LとYB-1の結合を促進し、3つのRNA結合タンパクが協調してエクソン10のスキッピングを誘導する。 |
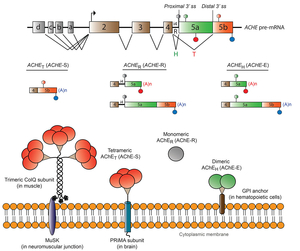 図27. アセチルコリンエステラーゼ遺伝子(ACHE)の選択的スプライシングとAChEアイソフォームの膜局在。 |
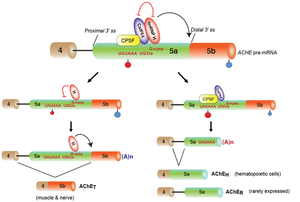 図28. hnRNP H結合による遠位3’スプライスサイト活性化によるAChETの産生とCstF64結合による選択的ポリアデニレーション部位(UAUAAA)活性化によるAChEHとAChERの産生。選択的ポリアデニレーション部位(pA-1)を赤丸で、恒常的ポリアデニレーション部位を青丸で示す。HnRNP Hが結合するG-runsとCstF64が結合するUG/UsはオーバーラップしておりhnRNP HとCstF64の競合によりスプライシングアイソフォームが決定する。 |
iv) エクソンアレイとRNA-seqによる網羅的なスプライシング解析
我々は正常ならびに病態におけるスプライシング解析のためにエクソンアレイとRNA-seqの両者を用いている。筋強直性ジストロフィー1型(DM1)の骨格筋におけるスプラシング異常を解析する目的でエクソンアレイ解析ツールを作成した。エクソンアレイ解析で得られるZ-scoreが選択的スプライシングならびに病的スプライシングを検出する最適の指標であることを見いだし、エクソンアレイ解析ツールを作成した(図29) (J Hum Genet 57: 368, 2012)。またエクソンアレイとRNA-seqの比較によりエクソンアレイがRNA-seqよりも選択的ならびに病的スプライシングを正確に検出できること見いだしている。両者の差異は、RNA-seqのみにPCRのステップが含まれていることが原因と想定される。
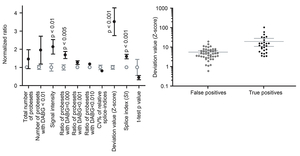 図29. Affymetrix exon array解析においてZ値がfalse positivesとtrue positivesを最も鋭敏に分別する。 |
v) CLIP-seqを含むマルチオミクス解析によるRNA結合タンパクの役割の解明
RNA結合タンパクMBNL1とCUGBP1は筋強直性ジストロフィー1型(DM1)の原因分子である。我々はMBNL1とCUGPB1のRNA結合部位をCLIP-seqにより同定し結合もチーフを明らかにした(図30)。加えて、結合部位特異的なスプライシング制御の存在を明らかにした(図31) (Sci Rep 2: 209, 2012)。さらに、両RNA結合タンパクは標的RNAの3’ UTRに結合し、標的RNAの分解を促進することを見いだした(図32)。
同様に筋萎縮性側索硬化症(ALS)と前頭側頭葉萎縮症(FTLD)の原因となるRNA結合タンパクFUSのRNA結合標的をCLIP-seqにより解析を行った。FUSはlong noncoding RNAであるPROMPTs (promoter upstream transcripts)に結合し標的遺伝子の発現を抑制することを見いだした(図33) (Sci Rep 2: 529, 2012)。CLIP-seq, ChIP-seq, RNA-seq, CAGE-seq, polyA-seqによるマルチオミクス解析とFUS標的遺伝子群の詳細な解析により、FUSはRNA転写産物(nascent RNA)に結合しRNAポリメラーゼIIを止めることを明らかにした。FUSがnascent RNA上の選択的ポリAサイトの下流に結合する時にはCPSF160を活性化しポリA付加を促進し3’末端が短縮した短いmRNAを作る。一方、FUSが 選択的ポリAサイトの上流に結合する時にはCPSF160の活性化は誘導されず短い転写産物は破壊される(図34) (Genes Dev 29: 1045, 2015)。
現在、スモールスケールのサンプルにおいてRNA結合タンパクの標的RNAの解明を行うための新規CLIP-seq手法の開発を行っており、RNA結合タンパクの機能のさらなる解明を行っている。
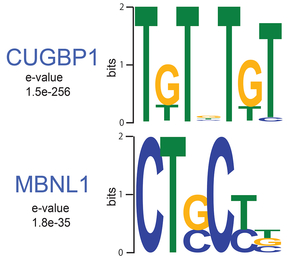 図30. CUGBP1とMBNL1のRNA結合モチーフ。 |
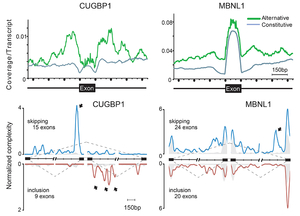 図31. CUGBP1とMBNL1は結合部位特異的にスプライシングを制御する。 |
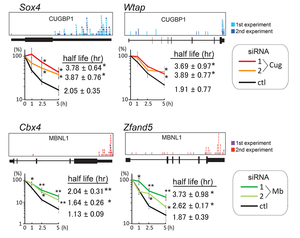 図32. CUGBP1とMBNL1は標的転写産物の3’ UTRsに結合しmRNAの崩壊を促進する。 |
 図33. FUSはプロモータ領域のアンチセンス鎖ノンコーディングRNA (PROMPTs)に結合し遺伝子発現を抑制する。 |
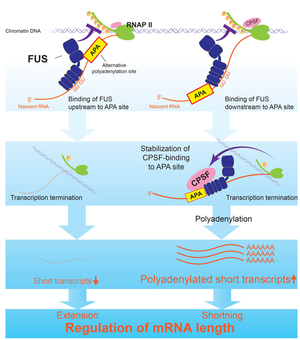 図34. FUSは結合部位特異的に3’末端を制御しmRNAの長さを決定する。 |
vi) siRNA設計アルゴリズム
マシーンラーニング手法により独自のsiRNA設計アルゴリズムiScoreを開発しウェブツールを提供している(図35, 36) (Nucleic Acids Res 35: e123, 2007)。iScore,は8種類のsiRNA設計アルゴリズムによるパラメータを同時に計算することにより、約50%の確率で高精度のsiRNAの設計が可能になる(図37)。単一のパラメータを計算するウェブツールでは約30%のヒット確率であるのに対して高い確度でsiRNAの設計を可能にしている。
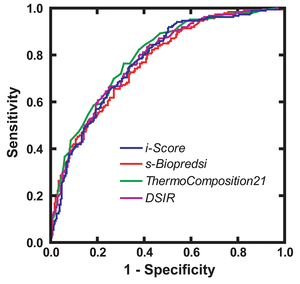 図35. siRNA設計ツールiScoreの感度・特異度は3種類の第2世代siRNA設計ツールに比肩する。 |
 図36. 熱不安定性があるsiRNAはすぐれたROCカーブを描き効果の予測精度が高い。 |
 図37. siRNA設計ウェブツールiScore。 |
3. 希少疾患に対するドラッグリポジショニング戦略
i) 神経筋疾患に対するドラッグリポジショニング
消炎鎮痛薬アスピリンの抗血小板作用、βアドレナリン受容体阻害剤プロプラノロールの震戦改善作用、睡眠薬サリドマイドの抗腫瘍効果、抗てんかん薬ゾニサミドの抗パーキンソン病作用など、既認可薬には予期せぬ薬効が存在する。既認可薬は至適投与量・最大投与量・至適投与方法・安全域・副作用が知られているため、モデル細胞・モデル動物で得た成果の臨床応用の敷居が低い。
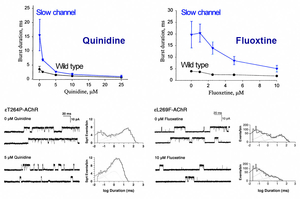
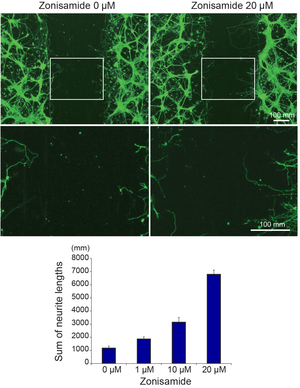
スローチャンネル症候群はアセチルコリン受容体(AChR)のチャンネル開口時間が異常に延長した病態である。我々は、抗不整脈剤キニジン(Neurology 60: 1710, 2003)と抗うつ薬SSRIフルオキセチン(Ann Neurol 43: 480, 1998)が臨床的に達成可能な濃度で、異常延長したAChRチャンネル開口時間を短縮することを明らかにした(図38)。また、抗てんかん薬・抗パーキンソン病薬ゾニサミドがNSC34細胞の神経突起を延長させることを見出し、マウス初代脊髄前角細胞でも濃度依存性の効果を確認した(図39)。ゾニサミドはマウス座骨神経のオートグラフトモデルに対して軸索再生を促進するとともに運動機能を改善させた(図40) (PLoS One 10: e0142786, 2015)。加えて、ゾニサミドは運動神経軸索スプラウティング促進作用により先天性筋無力症候群モデルマウスに対しても効果を発揮することを見出している。
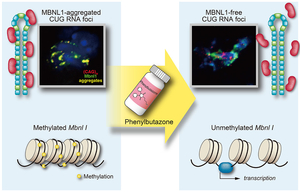
筋強直性ジストロフィー1型(DM1)はDMPK遺伝子3’非翻訳領域のCUG繰り返し配列の異常延長が原因である。RNAに転写された異常延長CUG繰り返し配列にRNA結合タンパクMBNL1が結合し、核内凝集体(nuclear foci)を作る。我々はMBNL1の発現誘導スクリーニングにより消炎鎮痛剤フェニルブタゾン(PBZ)がC2C12マウス筋芽細胞ならびにDM1モデルマウスにおいてMBNL1の発現を誘導することを見出した。DM1モデルマウス骨格筋においてPBZはClcn1, Nfix, Rpn2のスプライシング異常を改善し、骨格筋クロライドチャンネルの発現を上昇させ、筋中心核を減少させ、自発運動量を改善させた。PBZはMbnl1遺伝子イントロン1のメチル化を抑制し、MBNL1の異常延長CUG繰り返し配列への凝集をin vitroで抑制した(図41) (Sci Rep 6: 25317, 2016)。PBZは古い消炎鎮痛薬で副作用が多く、我々はより副作用が少ない薬剤の開発を行っている。
|
図38. 抗不整脈薬キニジンと抗うつ薬フルオキセチンはスローチャンネル症候群の異常延長したチャンネル開口時間を正常化する。 |
図39. マウス脊髄前角細胞の神経突起スクラッチアッセイにおいてゾニサミドは神経再生を促進する。 |
図40. ゾニサミドは抗てんかん薬として開発され、抗パーキンソン病薬としてリポジショニングされた。ゾニサミドは末梢神経障害、ニューロパチー、神経筋接合部障害への再度のリポジショニングが可能である可能性がある。 |
図41. 消炎鎮痛薬フェニルブタゾンは筋強直性ジストロフィーモデル1型モデルマウスの運動能を改善する。2つの作用機構があり、ひとつは異常延長CUG繰り返し配列へのMNBL1タンパクの結合阻害であり、もうひとつはMbnl1イントロン1の脱メチル化によるMbnl1発現誘導である。 |
ii) 骨・軟骨疾患に対するドラッグリポジショニング
進行性骨化性線維異形成症(FOP)に対して、カルシウムチャンネルブロッカーのうちオーストラリアで承認をされているperhexilineとドイツで承認されているfendilineが有効であることをモデル細胞とモデルマウスで検証した(図42) (J Bone Miner Metab 31: 26, 2013)。5名のFOP患者に対してパーヘキシリンのオープンラベル試験を行ったが有効性を確認できなかった(Orphanet J Rare Dis 8: 163, 2013)。我々はFOPに対してさらなる既認可薬の同定を進めている。
プロトンポンプ阻害剤ランソプラゾールは骨芽細胞のマスターレギュレータRunx2の核内移行を促進し骨芽細胞分化を促進することを見出した(図43)。ラットの大腿骨骨折モデルに対してランソプラゾールの経口投与は骨折治癒を促進した。ランソプラゾールはBMP/TGF-βによって活性化されるTAK1-p38 MAPK経路を活性化した。ランソプラゾールは培養細胞においてTRAF6のユビキチン化を促進し、その効果は脱ユビキチン化酵素CYLDの抑制によることを同定した。CYLDの構造モデリングと部位特異的アミノ酸変異導入にて、ランソプラゾールはCYLDに結合したユビキチンのC末端部分に存在するポケットに結合し、ユビキチンC末端のCYLDの酵素活性中心へのアクセスを阻害することを明らかにした(図44)。ランソプラゾールは骨芽細胞分化を促進する新規治療薬となり得る可能性がありさらなる開発をすすめている(図45) (EBioMedicine 2: 2046, 2015)。
軟骨無形成症はFGFR3の機能獲得変異が原因である。抗ヒスタミン薬・乗り物酔い薬メクロジンは軟骨細胞の増殖を促進し、FGF2を付加したRCS (rat chondrosarcoma)細胞の細胞外マトリックスの発現を誘導した。メクロジンは正常脛骨ならびにFGF2付加脛骨のexplant培養下での長軸方向への成長を促進するとともに(図46) (PLoS One 8: e81569, 2013)、軟骨無形成症モデルマウスの骨形成を改善した(図47) (Endocrinology 156: 548, 2015)。
カルシウムチャンネルブロッカーverapamilは変形性関節症(OA)患者軟骨細胞において可溶性のWnt阻害分子であるFRZBの発現を誘導することを見出したOAモデルラットに対してverapamilの関節内注射はOAの進行を抑制し、βカテニンの軟骨細胞核内移行を抑制した(図48) (PLoS One 9: e92699, 2014)。現在、他の骨軟骨疾患に対してもドラッグリポジショニング研究を精力的に行っている。
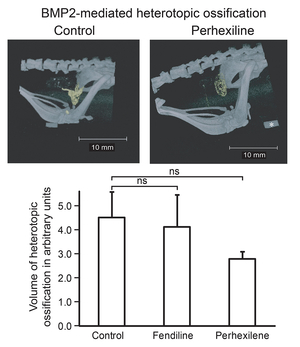 図42. 臨床で使われているカルシウムチャンネルブロッカー perhexilineとfendilineはFOPモデルマウスにおける異所性の骨格筋骨化を抑制する。 |
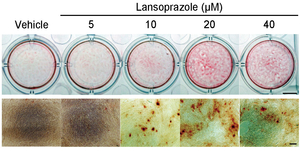 図43. 骨芽細胞への分化誘導をしたヒト骨髄由来間葉系幹細胞においてプロトンポンプ阻害薬ランソプラゾールはマトリックスカルシウム沈着を増強する。 |
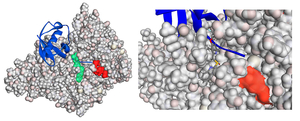 図44. CYLD (PDB id: 2VHF)へのユビキチン(青) (PDB id: 1NBF)結合のシムレーション. ユビキチンC末端が酵素活性中心(赤)に伸びている。ランソプラゾール(緑)はユビキチンC末端を横切るようにCYLDのポケットにフィットする。ポケット部位のR758とF766のアラニンへの置換(マークなし)はCYLD活性を損なうことなくランソプラゾールの反応性をなくす。 |
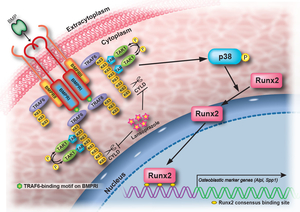 図45. ランソプラゾールによるRunx2活性化のモデル。 |
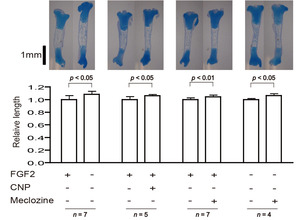 図46. 抗ヒスタミン薬メクロジンはFGF2存在下・非存在下におけるexplant培養をしたマウス胎児脛骨の成長を促進する。 |
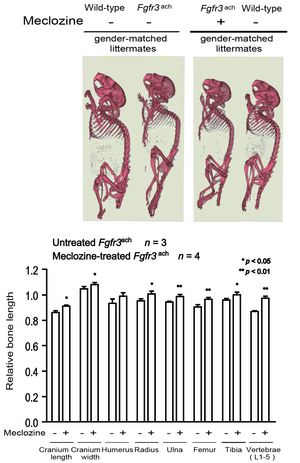 図47. メクロジンはFgfr3ach軟骨無形成症モデルマウスの骨成長を促進する |
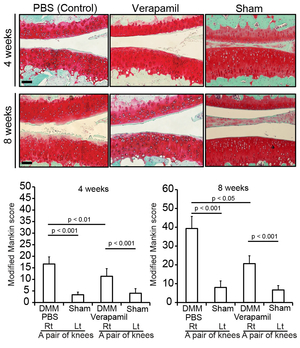 図48. 変形性関節症モデルラットへのカルシウム拮抗薬ベラパミルの関節内投与は変形性関節症の進行が軽減させる。 |
4. 腸内細菌叢解析によるパーキンソン病の病態機構解明
パーキンソン病(PD)は中脳黒質ならびに他の中枢神経領域におけるαシヌクレインの異常蓄積を特徴とする神経変性疾患である。αシヌクレイン異常蓄積は腸管神経叢から始まり迷走神経背側核、青斑核、中脳黒質へと上行することがほぼ確立している(図49)。PD患者においては腸管透過性が亢進し、腸管壁の大腸菌染色性・ニトロチロシン染色性が亢進している。我々は52名のPD患者と36名の同居健常人の腸内細菌叢解析を行いPDにおける腸内細菌叢の変化を報告した(PLoS One 10: e0142164, 2015)。機械学習によるモデル解析によりLactobacillus gasseriが進行したPDで多く、Clostridium coccoidesが初期のPDで多いことが判明した(図50)。加えて、水素産生菌がPDでは少なかった(図51)。我々はさらにPD患者と同居健常人の協力を得てショットガンメタゲノム解析ならびにメタボローム解析を開始している。
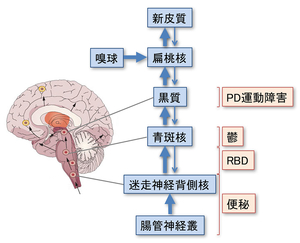 図49. Braakの報告による異常蓄積α-synucleinの迷走神経背側核から青斑核、中脳黒質への上行。RBD, REM睡眠行動障害。 |
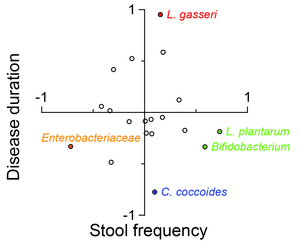 図50. パーキンソン病患者の腸内細菌叢のモデリング解析にてパーキンソン病の進行に関与する細菌と便秘の重症度に関与する細菌を明らかにした。 |
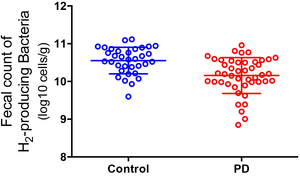 図51. パーキンソン病の腸内細菌叢には水素産生菌数が少ない。 |
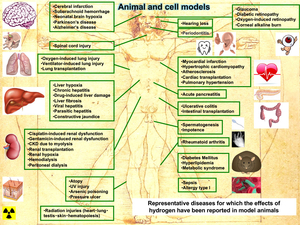
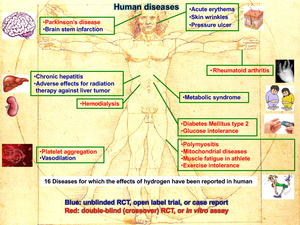
5. 分子状水素の酸化ストレス病態を中心とする多彩な疾患に対する効果とその分子作用機構解明
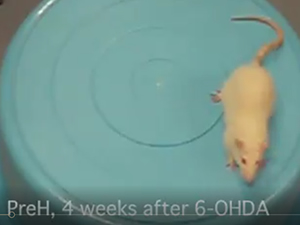
分子状水素の多彩な疾患モデル動物(図52)やヒト疾患(図53)における有効性が報告されてきた(Oxid Med Cell Longev 2012: 353152, 2012)。水素の効果は2007年から2015年までの9年間に321報の原著論文に報告されている(Med Gas Res 5: 12, 2015)。約3/4はマウス・ラットにおける効果の報告であるが、臨床研究も年々に増加しつつある。水素の効果はほとんどの臓器で報告さており、166の疾患モデル・ヒト疾患からなる31の病態で効果が報告されており、特に酸化ストレス関連病態ならびに炎症病で有効である。我々は、水素の効果をパーキンソン病モデルラット(図54, 55) (Neurosci Lett 453: 81, 2009)、ヒト炎症性筋疾患・ミトコンドリア病(Med Gas Res 1: 24, 2011)、子宮内虚血再灌流による新生児ラットの海馬障害(Free Radic Biol Med 69: 324, 2014)、ラット肺高血圧症(J Thorac Cardiovasc Surg 150: 645, 2015)、デュシャンヌ型筋ジストロフィーモデルmdxマウス(Redox Rep 1, 2016)、子宮内炎症による周産期脳障害(Free Radic Biol Med 91: 154, 2016)、ラット新生児の気管支肺異形成症(BPD) (Pediatr Pulmonol 51: 928, 2016)において効果を検証してきた。
水酸化ラジカルとパーオキシナイトライトの水素による特異的な除去が作用機構として最初に提唱されたが、ラジカル除去効果のみでは水素の顕著な効果を説明できない。我々は水素がFcεRIを介した肥満細胞のシグナル伝達系を抑制することを報告した(Biochem Biophys Res Commun 389: 651, 2009)。さらに、水素はマクロファージにおいてLPS/インターフェロンγ誘発による一酸化窒素産生を抑制することを報告した(Biochem Biophys Res Commun 411: 143, 2011)。これらは水素のラジカル除去効果では説明ができない。遺伝子発現の網羅的な解析により水素は複数のシグナル経路を変容させることを明らかにした(Mol Cell Biochem 403: 231, 2015)。さらに、水素はβカテニン破壊複合体のリン酸化活性を促進することによりWnt/βカテニンシグナル伝達系を抑制 することを明らかにした(Sci Rep 6: 31986, 2016)。加えて、水素水と間欠水素ガス吸入がパーキンソン病モデルラットに有効である一方、持続的な水素ガス吸入は無効であることを報告した(Med Gas Res 2: 15, 2012)。現在、水素の幅広い効果を説明できる単一の標的分子同定に向けて精力的に研究を行っている。
|
図52. 分子状水素が有効であることが報告された代表的な疾患モデル。 |
図53. 分子状水素が有効であることが報告された代表的なヒト疾患。 |
|
|
図54. コントール水を自由飲水した片側パーキンソン病モデルラットはアンフェタミン腹腔内投与後激しく回転する。[movie] |
|

教員
| 構成員名 | 役職 | 所属 |
|---|---|---|
| 増田 章男 | 准教授 | 神経遺伝情報学 |
| 伊藤 美佳子 | 講師 | 神経遺伝情報学 |
| 濵口 知成 | 特任講師 | 学術研究・産学官連携推進本部メディカルイノベーション推進室 |
| 西脇 寛 | 助教 | 神経遺伝情報学 |
研究実績
- 2023年
- Kawashima I, Matsushita M, Mishima K, Kamiya Y, Osawa Y, Ohkawara B, Ohno K, Kitoh H, Imagama S. Activated fgfr3 suppresses bone regeneration and bone mineralization in an ovariectomized mouse model. BMC Musculoskelet Disord 2023, 24: 200.
- Zhang S, Ohkawara B, Ito M, Huang Z, Zhao F, Nakata T, Takeuchi T, Sakurai H, Komaki H, Kamon M, Araki T, and Ohno K. A mutation in DOK7 in congenital myasthenic syndrome forms aggresome in cultured cells, and reduces DOK7 expression and MuSK phosphorylation in patient-derived iPS cells. Hum Mol Genet 2023, 32: 1511-1523.
- Kamiya Y, Matsushita M, Mishima K, Ohkawara B, Michigami T, Imagama S, Ohno K, Kitoh H. Meclozine ameliorates bone mineralization and growth plate structure in a mouse model of X‑linked hypophosphatemia. Exp Ther Med 2023, 25: 39.
- Shen XM, Nakata T, Mizuno S, Imoto I, Selcen D, Ohno K, Engel AG. Impaired gating of γ- and ε-AChR respectively causes Escobar syndrome and fast channel myasthenia. Ann Clin Transl Neurol 2023, 10: 732-43.
- Yamashita Y, Nakada S, Nakamura K, Sakurai H, Ohno K, Goto T, Mabuchi Y, Akazawa C, Hattori N, Arikawa-Hirasawa E. Evaluation of Human-Induced Pluripotent Stem Cells Derived from a Patient with Schwartz–Jampel Syndrome Revealed Distinct Hyperexcitability in the Skeletal Muscles. Biomedicines, 2023, 11: 814.
- Fuse Y, Nishiwaki H, Imaizumi T, Nagata Y, Ohno K, Saito R. Machine learning models predict delayed hyponatremia post-transsphenoidal surgery using clinically available features. Pituitary, 2023, 26: 237-249.
- Bushra S*, Lin Y*, Joudaki A, Ito M, Ohkawara B, Ohno K, Masuda A. Neural isoforms of agrin are generated by reduced PTBP1−RNA interaction network spanning the neuron-specific splicing regions in AGRN. Int J Mol Sci 2023, 24: 7420. *Equal contributions.
- Muraoka A, Suzuki M, Hamaguchi T, Watanabe S, Iijima K, Murofushi Y, Shinjo K, Osuka S, Hariyama Y, Ito M, Ohno K, Kiyono T, Kyo S, Iwase A, Kikkawa F, Kajiyama H, Kondo Y. Fusobacterium infection facilitates the development of endometriosis through the phenotypic transition of endometrial fibroblasts. Sci Trans Med 2023, 5: eadd1531.
- Farshadyeganeh P*, Nazim M*, Zhang R, Ohkawara B, Nakajima K, Rahman MA, Nasrin F, Ito M, Takeda J, Ohe K, Miyasaka Y, Ohno T, Masuda A, Ohno K. Splicing regulation of GFPT1 muscle-specific isoform and its roles in glucose metabolisms and neuromuscular junction. iScience 2023, 26: 107746. *Equal contributions.
- Joudaki A, Takeda J, Masuda A, Ode R, Fujiwara K, Ohno K. FexSplice: A LightGBM-Based Model for Predicting the Splicing Effect of a Single Nucleotide Variant Affecting the First Nucleotide G of an Exon. Genes 2023, 14: 1765.
- Huang Z, Ito M, Zhang S, Toda T, Takeda J, Ogi T, Ohno K. Extremely low-frequency electromagnetic field induces acetylation of heat shock proteins and enhances protein folding. Ecotox Environ Safe 2023, 264: 115482.
- Fuse Y, Nagashima Y, Nishiwaki H, Muramatsu Y, Ohka F, Araki Y, Nishimura Y, Watanabe K, Ohno K, Saito R. Development of machine learning models for predicting unfavorable functional outcomes from preoperative data in patients with chronic subdural hematomas. Sci Rep in press.
- Ohno K, Ohkawara B, Shen X-M, Selcen D, Engel AG. Clinical and pathologic features of congenital myasthenic syndromes caused by 35 genes – a comprehensive review. Int J Mol Sci 2023; 24: 3730 (査読有)
- LeBaron TW, Ohno K, Hancock JT. The On/off History of Hydrogen in Medicine: Will the interest persist this time around? Oxygen 2023, 3: 143-162 (査読有)
- Hirayama M, Nishiwaki H, Hamaguchi T, Ohno K. Gastrointestinal disorders in parkinson's disease and other lewy body diseases. npj Parkinsons Dis 2023, 9: 71. (査読有)
- Hirayama M, Ohno K. Gut Microbiota Changes and Parkinson’s Disease: What Do We Know, Which Avenues Ahead. Gut Microbiota in Aging and Chronic Diseases Ed. by Francesco Marotta Healthy Ageing and Longevity 17 Editor-in-Chief: Suresh I. S. Rattan. Springer Nature, Switzerland, 2023, pp. 257-278 (査読有)
- 2022年
- Tanaka H, Matsumura S, Ishikawa K, Hashizume H, Ito M, Nakamura K, Kajiyama H, Kikkawa F, Ito M, Ohno K, Okazaki Y, Toyokuni S, Mizuno M, Hori M. Enhancement of ethanol production and cell growth in budding yeast by direct irradiation of low-temperature plasma. Jpn J Appl Phys 2022, 61: SA1007.
- Sakaguchi T, Miyamoto K, Ohkawara B, Kisimoto Y, Ishizuka S, Hiraiwa H, Imagama S, Ishiguro N, Ohno K. Promethazine downregulates Wnt/β-catenin signaling and increases biomechanical forces of injured Achilles tendon in early stage of healing. Am J Sport Med 2022, 50: 1317-27.
- Takahashi K, Nishiwaki H, Ito M, Iwaoka K, Takahashi K, Suzuki Y, Taguchi K, Yamahara K, Tsuboi Y, Kashihara K, Hirayama M, Ohno K, Maeda T. Altered gut microbiota in Parkinson's disease patients with motor complications. Parkinsonism Relat Disord 2022, 95: 11-17.
- Kawamura Y, Hida T, Ohkawara B, Matsushita M, Kobayashi T, Ishizuka S, Hiraiwa H, Tanaka S, Tsushima M, Nakashima H, Ito K, Imagama S, Ito M, Masuda A, Ishiguro N, Ohno K. Meclozine ameliorates skeletal muscle pathology and increases muscle forces in mdx mice. Biochem Biophys Res Commun 2022, 592: 87-92.
- Hasegawa T, Ito M, Hasegawa S, Teranishi M, Takeda K, Negishi S, Nishiwaki H, Takeda J, LeBaron TW, Ohno K. Molecular hydrogen enhances proliferation of cancer cells that exhibit potent mitochondrial unfolded protein response. Int J Mol Sci 2022, 23: 2888.
- Koike H, Nishida Y, Shinomura T, Ohkawara B, Ohno K, Zhuo Lisheng, Kimata K, Ushida T, Imagama S. Possible repositioning of an oral anti-osteoporotic drug, Ipriflavone, for treatment of inflammatory arthritis via inhibitory activity of KIAA1199, a novel potent hyaluronidase. Int J Mol Sci 2022, 23, 4089
- Nishiwaki H, Ito M, Hamaguchi T, Maeda T, Kashihara K, Tsuboi Y, Ueyama J, Yoshida T, Hanada H, Takeuchi I, Katsuno M, Hirayama M, Ohno K. Short chain fatty acids-producing and mucin-degrading intestinal bacteria predict the progression of early Parkinson’s disease. npj Parkinsons Dis 2022, 8: 65.
- Toda T, Ito M Takeda J, Masuda A, Mino H, Hattori N, Mohri K, Ohno K. Extremely low-frequency pulses of faint magnetic field induce mitophagy to rejuvenate mitochondria. Commun Biol 2022, 5: 453.
- Ito K, Nishida Y, Hamada S, Shimizu K, Sakai T, Ohkawara B, Alman BA, Enomoto A, Ikuta K, Koike H, Zhang J, Ohno K, Imagama S. Efficacy of auranofin as an inhibitor of desmoid progression. Sci Rep 12: 11918, 2022.
- Gibo N, Hamaguchi T, Miki Y, Yamamura T, Nakaguro M, Ito M, Nakamuara M, Kawashima H, Hirayama M, Hirooka Y, Wakabayashi K, Ohno K. Examination of abnormal alpha-synuclein aggregates in the enteric neural plexus in patients with ulcerative colitis. J Gastrointestin Liver Dis 2022, 31: 290-300.
- Nishiwaki H, Ueyama J, Kashihara K, Ito M, Hamaguchi T, Maeda T, Tsuboi Y, Katsuno M, Hirayama M, Ohno K. Gut microbiota in dementia with Lewy bodies. npj Parkinsons Dis 2022, 8: 169.
- Ueyama J, Hayashi M, Hirayama M, Nishiwaki H, Ito M, Saito I, Tsuboi Y, Isobe T, Ohno K. Effects of pesticide intake on gut microbiota and metabolites in healthy adults. Int J Environ Res Public Health 2022, 20: 213.
- Mishima K, Okabe YT, Mizuno M, Ohno K, Kitoh H, Imagama S, Efficacy of soluble lansoprazole-impregnated beta-tricalcium phosphate for bone regeneration. Sci Rep 2022, 12: 20550.
- Hirayama M, Ohno K. Reply to the Letter to the Editor “The microbiota in Parkinson's disease: ranking the risk of heart disease”. Ann Nutr Metab 2022; 78: 119-120 (査読有)
- LeBaron TW, Sharpe R, Ohno K. Electrolyzed Reduced Water: Review I. Molecular hydrogen is the Exclusive Agent Responsible for the Therapeutic Effects. Int J Mol Sci 2022; 23(23): 14750 (査読有)
- LeBaron TW, Sharpe R, Ohno K. Electrolyzed Reduced Water: Review II. Safety Concerns and Effectiveness as a Source of Hydrogen Water. Int J Mol Sci 2022; 23(23): 14508 (査読有)
- 2021年
- Kusano T, Nakatani M, Ishiguro N, Ohno K, Yamamoto N, Morita M, Yamada H, Uezumi A, Tsuchida K. Desloratadine inhibits heterotopic ossification by suppression of BMP2-Smad1/5/8 signaling. J Orthop Res 2021, 39: 1297–1304.
- Tawara N, Yamashita S, Takamatsu K, Yamasaki Y, Mukaino A, Nakane S, Farshadyeganeh P, Ohno K, Ando Y. Efficacy of salbutamol monotherapy in slow-channel congenital myasthenic syndrome caused by a novel mutation in CHRND. Muscle Nerve 2021, 63: E30-E32.
- Abe K, Hirayama M, Ohno K, Shimamura T. Hierarchical non-negative matrix factorization using clinical information for microbial communities. BMC Genomics 2021, 22: 104.
- Inoue T, Ohkawara B, Bushra S, Kanbara S, Nakashima H, Koshimizu H, Tomita H, Ito M, Masuda A, Ishiguro N, Imagama S, Ohno K. Zonisamide upregulates neuregulin-1 expression and enhances acetylcholine receptor clustering at the in vitro neuromuscular junction. Neuropharmacology 2021, 195: 108637.
- Takeda J*, Fukami S*, Tamura A, Shibata A, Ohno K. Intsplice2: Prediction of the splicing effects of intronic single-nucleotide variants using lightGBM modeling. Front Genet 2021, 12: 701076. *Equal contributions.
- Tanaka H, Maeda S, Nakamura K, Hashizume H, Ishikawa K, Ito M, Ohno K, Mizuno M, Motooka Y, Okazaki Y, Toyokuni S, Kajiyama H, Kikkawa F, Hori M. Plasma-activated Ringer’s lactate solution inhibits cellular respiratory system in HeLa cells. Plasma Processes and Polymers 2021, 18: e2100056.
- Kawachi T, Masuda A, Yamashita Y, Takeda J, Ohkawara B, Ito M, Ohno K. Regulated splicing of large exons is linked to phase-separation of vertebrate transcription factors. EMBO J 2021, 40(22): e107485.
- Hirayama M*, Nishiwaki H*, Hamaguchi T, Ito M, Ueyama J, Maeda T, Kashihara K, Tsuboi Y, Ohno K. Intestinal Collinsella may mitigate infection and exacerbation of COVID-19 by producing ursodeoxycholate. PLoS One 2021, 16: e0260451. *Equal contributions.
- Takemoto G, Matsushita M, Okamoto T, Ito T, Matsuura Y, Takashima C, Chen-Yoshikawa TF, Ebi H, Imagama S, Kitoh H, Ohno K, Hosono Y. Meclozine attenuates the MARK pathway in mammalian chondrocytes and ameliorates Fgf2-induced bone hyperossification in larval zebrafish. Front Cell Dev Biol 2021, 9: 694018.
- Ohkawara B, Ito M, Ohno K. Secreted signaling molecules at the neuromuscular junction in physiology and pathology. Int J Mol Sci 2021, 22: 2455 (査読有)
- Masuda A, Kawachi T, Ohno K. Rapidly growing protein-centric technologies to extensively identify protein-RNA interactions: Application to the analysis of co-transcriptional RNA processing. Int J Mol Sci 2021, 22: 5312. (査読有)
- Hirayama M, Ohno K. Parkinson’s disease and gut microbiota. Ann Nutr Metab 2021; 77 (Suppl 2):28-35. (査読有)
- 2020年
- Ozeki N, Yamawaki-Ogata A, Narita Y, Mii S, Ushida K, Ito M, Hirano S, Kurokawa R, Ohno K, Usui A. Hydrogen water alleviates obliterative airway disease in mice. Gen Thorac Cardiovasc Surg 2020, 68: 158-163.
- Ueyama J, Oda M, Hirayama M, Sugitate K, Sakui N, Hamada R, Ito M, Saito I, Ohno K. Freeze-drying enables homogeneous and stable sample preparation for determination of fecal short-chain fatty acids. Anal Biochem 2020, 589: 113508.
- Huang K, Masuda A, Chen G, Bushra S, Kamon M, Araki T, Kinoshita M, Ohkawara B, Ito M, Ohno K. Inhibition of cyclooxygenase-1 by nonsteroidal anti-inflammatory drugs demethylates MeR2 enhancer and promotes Mbnl1 transcription in myogenic cells. Sci Rep 2020, 10: 2558.
- Nakazawa Y, Hara Y, Oka Y, Komine O, van den Heuvel D, Guo C, Daigaku Y, Isono M, He Y, Shimada M, Kato K, Jia N, Hashimoto S, Kotani Y, Miyoshi Y, Tanaka M, Sobue A, Mitsutake N, Suganami T, Masuda A, Ohno K, Nakada S, Mashimo T, Yamanaka K, Luijsterburg MS, Ogi T. Ubiquitination of DNA Damage-Stalled RNAPII Promotes Transcription-Coupled Repair. Cell 2020, 180: 1228-1244 e24.
- Masuda A, Kawachi T, Takeda J, Ohkawara B, Ito M, Ohno K. tRIP-seq reveals repression of premature polyadenylation by co-transcriptional FUS-U1 snRNP assembly. EMBO Rep 2020, 21: e49890.
- Ohkawara B, Shen X, Selcen D, Nazim M, Bril V, Tarnopolsky MA, Brady L, Fukami S, Amato AA, Yis U, Ohno K, Engel AG. Congenital myasthenic syndrome-associated agrin variants affect clustering of acetylcholine receptors in a domain-specific manner. JCI Insight 2020, 5(7):e132023.
- Nishiwaki H, Ito M, Ishida T, Hamaguchi T, Maeda T, Kashihara K, Tsuboi Y, Ueyama J, Shimamura T, Mori H, Kurokawa K, Katsuno M, Hirayama M, Ohno K. Meta-Analysis of Gut Dysbiosis in Parkinson’s Disease. Mov Disord 2020, 35(9): 1626-1635.
- Takeda J, Nanatsue K, Yamagishi R, Ito M, Haga N, Hirata H, Ogi, Ohno K. InMeRF: prediction of pathogenicity of missense variants by individual modeling for each amino acid substitution. NAR Genomics and Bioinformatics 2020, 2(2): lqaa038.
- Ohkawara B*, Kobayakawa A*, Kanbara S, Hattori T, Kubota S, Ito M, Masuda A, Takigawa M, Lyons KM, Ishiguro N, Ohno K. CTGF/CCN2 facilitates LRP4-mediated formation of the embryonic neuromuscular junction. EMBO Rep 2020, 21(8): e48462. *Equal contribution
- Kanbara S, Ohkawara B, Nakashima H, Ohta K, Koshimizu H, Inoue T, Tomita H, Ito M, Masuda A, Ishiguro N, Imagama S, Ohno K. Zonisamide ameliorates progression of cervical spondylotic myelopathy in a rat model. Sci Rep 2020, 10: 13138.
- Huang K, Li J, Ito M, Takeda J, Ohkawara B, Ogi T, Masuda A, Ohno K. Gene Expression Profile at the Motor Endplate of the Neuromuscular Junction of Fast-Twitch Muscle. Front Mol Neurosci 2020, 13: 154.
- Nishiwaki H, Hamaguchi T, Ito M, Ishida T, Maeda T, Kashihara K, Tsuboi Y, Ueyama J, Shimamura T, Mori H, Kurokawa K, Katsuno M, Hirayama M, Ohno K. Short-chain fatty acid-producing gut microbiota is decreased in Parkinson's disease but not in rapid-eye-movement sleep behavior disorder. mSystems 2020, 5: e00797-00720.
- Takeuchi A, Takahashi Y, Iida K, Hosokawa M, Irie K, Ito M, Brown JB, Ohno K, Nakashima K, Hagiwara M. Identification of Qk as a Glial Precursor Cell Marker that Governs the Fate Specification of Neural Stem Cells to a Glial Cell Lineage. Stem. Stem Cell Rep 2020, 15: 883-897.
- Koshimizu H*, Ohkawara B*, Nakashima H, Ota K, Kanbara S, Inouse T, Tomita H, Sayo A, Kiryu-Seo S, Konishi H, Ito M, Masuda A, Ishiguro N, Imagama S, Kiyama H, Ohno K. Zonisamide ameliorates neuropathic pain by suppressing microglial activation in the spinal cord in a mouse model. Life Sci 2020, 263: 118577. *Equal contributions.
- 2019年
- Okura T, Ohkawara B, Takegami Y, Ito M, Masuda A, Seki T, Ishiguro N, Ohno K. Mianserin suppresses R-spondin 2-induced activation of Wnt/β-catenin signaling in chondrocytes and prevents cartilage degradation in a rat model of osteoarthritis. Sci Rep 2019, 9: 2808.
- Abe K, Hirayama M, Ohno K, Shimamura T. ENIGMA: an enterotype-like unigram mixture model for microbial association analysis. BMC Genomics 2019, 20: 191.
- Tsuda T, Nonome T, Goto S, Takeda J, Tsunoda M, Hirayama M, Ohno K. Application of Skin Gas GC/MS Analysis for Prediction of the Severity Scale of Parkinson’s Disease. Chromatography 2019, 40: 149-155.
- Kataoka N, Maeda A, Ohno K. RNA Diseases in Humans – From Fundamental Research to Therapeutic Applications. Front Mol Biosci, 2019, 6:53 (査読有)
- 2018年
- Yu Y, Lin Y, Takasaki Y, Wang C, Kimura H, Xing J, Ishizuka K, Toyama M, Kushima I, Mori D, Arioka Y, Uno Y, Shiino T, Nakamura Y, Okada T, Morikawa M, Ikeda M, Iwata N, Okahisa Y, Takaki M, Sakamoto S, Someya T, Egawa J, Usami M, Kodaira M, Yoshimi A, Oya-Ito T, Aleksic B, Ohno K, Ozaki N. Rare loss of function mutations in N-methyl-D-aspartate glutamate receptors and their contributions to schizophrenia susceptibility. Transl Psychiatry 2018, 8: 12.
- Kurahashi H, Azuma Y, Masuda A, Okuno T, Nakahara E, Imamura T, Saitoh M, Mizuguchi M, Shimizu T, Ohno K, Okumura A. MYRF is associated with encephalopathy with reversible myelin vacuolization. Ann Neurol 2018, 83: 98-106.
- Ito K*, Ohkawara B*, Yagi H, Nakashima H, Tsushima M, Ota K, Konishi H, Masuda A, Imagama S, Kiyama H, Ishiguro N, Ohno K. Lack of Fgf18 causes abnormal clustering of motor nerve terminals at the neuromuscular junction with reduced acetylcholine receptor clusters. Sci Rep 2018, 8: 434. *Equal contribution.
- Nishiwaki H, Ito M, Negishi S, Sobue S, Ichihara M, Ohno K. Molecular hydrogen upregulates heat shock response and collagen biosynthesis, and downregulates cell cycles: meta-analyses of gene expression profiles. Free Radic Res 2018, 52: 434-445.
- Takeuchi A, Iida K, Tsubota T, Hosokawa M, Denawa M, Brown JB, Ninomiya K, Ito M, Kimura H, Abe T, Kiyonari H, Ohno K, Hagiwara M. Loss of Sfpq causes long-gene transcriptopathy in the brain. Cell Rep 2018, 23: 1326-1341.
- Li J, Ito M, Ohkawara B, Masuda A, Ohno K. Differential effects of spinal motor neuron-derived and skeletal muscle-derived Rspo2 on acetylcholine receptor clustering at the neuromuscular junction. Sci Rep 2018, 8: 13577.
- Abe K, Hirayama M, Ohno K, Shimamura T. A latent allocation model for the analysis of microbial composition and disease. BMC Bioinformatics 2018, 19: 519.
- Hirayama M, Ito M, Minato T, Yoritaka A, LeBaron TW, Ohno K. Inhalation of hydrogen gas elevates urinary 8-hydroxy-2'-deoxyguanine in Parkinson's disease. Med Gas Res 2018, 8: 144-149.
- Suzuki A, Ito M, Hamaguchi T, Mori H, Takeda Y, Baba R, Watanabe T, Kurokawa K, Asakawa S, Hirayama M, Ohno K. Quantification of hydrogen production by intestinal bacteria that are specifically dysregulated in Parkinson's disease. PLoS One 2018, 13: e0208313.
- Yoritaka A, Ohtsuka C, Maeda T, Hirayama M, Abe T, Watanabe H, Saiki H, Oyama G, Fukae J, Shimo Y, Hatano T, Kawajiri S, Okuma Y, Machida Y, Miwa H, Suzuki C, Kazama A, Tomiyama M, Kihara T, Hirasawa M, Shimura H, Oda E, Ito M, Ohno K, Hattori N. Randomized, double-blind, multicenter trial of hydrogen water for Parkinson's disease. Mov Disord 2018, 33: 1505-1507.
- Ohno K, Takeda JI, Masuda A. Rules and tools to predict the splicing effects of exonic and intronic mutations. Wiley Interdiscip Rev RNA 2018, 9: e1451. (査読有)
- Ito M, Ohno K. Protein-anchoring therapy to target extracellular matrix proteins to their physiological destinations. Matrix Biol, 2018, 68-69: 628-636. (査読有)
- 2017年
- Nazim M, Masuda A, Rahman MA, Nasrin F, Takeda JI, Ohe K, Ohkawara B, Ito M, Ohno K. Competitive regulation of alternative splicing and alternative polyadenylation by hnRNP H and CstF64 determines acetylcholinesterase isoforms. Nucleic Acids Res 2017, 45: 1455-1468.
- Hasegawa S, Ito M, Fukami M, Hashimoto M, Hirayama M, Ohno K. Molecular hydrogen alleviates motor deficits and muscle degeneration in mdx mice. Redox Rep 2017, 22: 26-34.
- Matsushita M, Mishima K, Esaki R, Ishiguro N, Ohno K, Kitoh H. Maternal administration of meclozine for the treatment of foramen magnum stenosis in transgenic mice with achondroplasia. J Neurosurg Pediatr 2017, 19: 91-95.
- Ito M*, Ehara Y*, Li J, Inada K, Ohno K. Protein-anchoring therapy of biglycan for mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther 2017, 28: 428-436. *Equal contribution.
- Takeda JI, Masuda A, Ohno K. Six GU-rich (6GUR) FUS-binding motifs detected by normalization of CLIP-seq by Nascent-seq. Gene 2017, 518: 57-64.
- Kishimoto Y, Ohkawara B, Sakai T, Ito M, Masuda A, Ishiguro N, Shukunami C, Docheva D, Ohno K. Wnt/β-catenin signaling suppresses expressions of Scx, Mkx, and Tnmd in tendon-derived cells. PLoS One, 2017, 12: e0182051.
- Ahsan KB, Masuda A, Rahman MA, Takeda J, Nazim M, Ohkawara B, Ito M, Ohno K. SRSF1 suppresses selection of intron-distal 5’ splice site of DOK7 intron 4 to generate functional full-length Dok-7 protein. Sci Rep 2017, 7: 10446.
- Tabeta K, Du X, Arimatsu K, Yokoji M, Takahashi N, Amizuka N, Hasegawa T, Crozat K, Maekawa T, Miyauchi S, Matsuda Y, Ida T, Kaku M, Hoebe K, Ohno K, Yoshie H, Yamazaki K, Moresco EM, Beutler B. An ENU-induced splice site mutation of mouse Col1a1 causing recessive osteogenesis imperfecta and revealing a novel splicing rescue. Sci Rep 2017, 7: 11717.
- Miyamoto K, Ohkawra B, Ito M, Masuda A, Hirakawa A, Sakai T, Hiraiwa H, Hamada T, Ishiguro N, Ohno K. Fluoxetine ameliorates cartilage degradation in osteoarthritis by inhibiting Wnt/β-catenin signaling. PLoS One 2017, 12: e0184388.
- Minato T, Maeda T, Fujisawa Y, Tsuji H, Nomoto K, Ohno K, Hirayama M. Progression of Parkinson's disease is associated with gut dysbiosis: Two-year follow-up study. PLoS One 2017, 12: e0187307.
- Gao KP, Ren YC, Wang JJ, Liu ZC, Li JN, Li LL, Wang BY, Li H, Wang YX, Cao YK, Ohno K, Zhai RH, Liang Z. Interactions between genetic polymorphisms of glucose metabolizing genes and smoking and alcohol consumption in the risk of type 2 diabetes mellitus. Appl Physiol Nutr Med 2017, 42: 1316-1321.
- Kasai T, Nakatani M, Ishiguro N, Ohno K, Yamamoto N, Morita M, Yamada H, Tsuchida K, Uezumi A. Promethazine hydrochloride inhibits ectopic fat cell formation in skeletal muscle. Am J Pathol 2017, 187: 2627-2634.
- Osawa Y, Matsushita M, Hasegawa S, Esaki R, Fujio M, Ohkawara B, Ishiguro N, Ohno K, Kitoh H. Activated FGFR3 promotes bone formation via accelerating endochondral ossification in mouse model of distraction osteogenesis. Bone 2017, 105: 42-49.
- Ohno K, Rahman MA, Nazim M, Nasrin F, Lin Y, Takeda JI, Masuda A. Splicing regulation and dysregulation of cholinergic genes expressed at the neuromuscular junction. J Neurochem 2017, 142 Suppl 2: 64-72. (査読有)
- Ohno K, Ohkawara B, Ito M. Agrin-LRP4-MuSK signaling as a therapeutic target for myasthenia gravis and other neuromuscular disorders. Expert Opin Ther Targets 2017, 21: 949-958.(査読有)
- 2016年
- Yagi H, Ohkawara B, Nakashima H, Ito K, Tsushima M, Ishii H, Noto K, Ohta K, Masuda M, Imagama S, Ishiguro N, Ohno K. Zonisamide enhances neurite elongation of primary motor neurons and facilitates peripheral nerve regeneration in vitro and in a mouse model. PLoS One 2016, 11: e0148470.
- Imai K, Kotani T, Tsuda H, Mano Y, Nakano T, Ushida T, Li H, Miki R, Sumigama S, Iwase A, Hirakawa A, Ohno K, Toyokuni S, Takeuchi H, Mizuno T, Suzumura A, Kikkawa F. Neuroprotective potential of molecular hydrogen against perinatal brain injury via suppression of activated microglia. Free Radic Biol Med 2016, 91: 154-163.
- Hasegawa S, Kitoh H, Ohkawara B, Mishima K, Matsushita M, Masuda A, Ishiguro N, Ohno K. Tranilast stimulates endochondral ossification by upregulating SOX9 and RUNX2 promoters. Biochem Biophys Res Commun, 2016, 470: 356-361.
- Gao K, Wang J, Li L, Zhai Y, Ren Y, You H, Wang B, Wu X, Li J, Liu Z, Li X, Huang Y, Luo XP, Hu D, Ohno K, Wang C. Polymorphisms in four genes (rs151290, rs972283, rs780094 and rs10830963) and their correlation with type 2 diabetes mellitus in Han Chinese in Henan Province, China. Int J Env Res Public Health 2016, 13.
- Takegami Y, Ohkawara B, Ito M, Masuda A, Nakashima H, Ishiguro N, Ohno K. R-spondin 2 facilitates differentiation of proliferating chondrocytes into hypertrophic chondrocytes by enhancing Wnt/beta-catenin signaling in endochondral ossification. Biochem Biophys Res Commun 2016, 473: 255-264.
- Chen G, Masuda A, Konishi H, Ohkawara B, Ito M, Kinoshita M, Kiyama H, Matsuura T, Ohno K. Phenylbutazone induces expression of MBNL1 and suppresses formation of MBNL1-CUG RNA foci in a mouse model of myotonic dystrophy. Sci Rep 2016, 6: 25317.
- Hirayama M, Tsunoda M, Yamamoto M, Tsuda T, Ohno K. Serum tyrosine-to-phenylalanine ratio is low in Parkinson's disease. J Parkinsons Dis 2016, 6: 423-431.
- Nakashima H*, Ohkawara B*, Ishigaki S, Fukudome T, Ito K, Tsushima M, Konishi H, Okuno T, Yoshimura T, Ito M, Masuda A, Sobue G, Kiyama H, Ishiguro N, Ohno K. R-spondin 2 promotes acetylcholine receptor clustering at the neuromuscular junction via Lgr5. Sci Rep 2016, 6: 28512. *Equal contribution.
- Bruun GH, Doktor TK, Borch J-J, Masuda A, Krainer AR, Ohno K, Andresen BS. Global identification of hnRNP A1 binding sites for SSO-based splicing modulation. BMC Biol 2016, 14: 54.
- Shibata A, Okuno T, Rahman MA, Azuma Y, Takeda J, Masuda A, Selcen D, Engel AG, Ohno K. IntSplice: prediction of the splicing consequences of intronic single-nucleotide variations in the human genome. J Hum Genet 2016, 61: 633-640.
- Muramatsu Y, Ito M, Oshima T, Kojima S, Ohno K. Hydrogen-rich water ameliorates bronchopulmonary dysplasia (BPD) in newborn rats. Pediatr Pulmonol 2016, 51: 928-935.
- Lin Y, Ohkawara B, Ito M, Misawa N, Miyamoto K, Takegami Y, Masuda A, Toyokuni S, Ohno K. Molecular hydrogen suppresses activated Wnt/beta-catenin signaling. Sci Rep 2016, 6: 31986.
- Shen X-M*, Okuno T*, Milone M, Otsuka K, Takahashi K, Komaki H, Giles E, Ohno K, Engel AG. Mutations causing slow-channel myasthenia reveal that a valine ring in the channel pore of muscle AChR is optimized for stabilizing channel gating. Hum Mutat, 2016, 37: 1051-1059. *Equal contribution.
- Ushida T, Kotani T, Tsuda H, Imai K, Nakano T, Hirako S, Ito Y, Li H, Mano Y, Wang J, Miki R, Yamamoto E, Iwase A, Bando YK, Hirayama M, Ohno K, Toyokuni S, Kikkawa F. Molecular hydrogen ameliorates several characteristics of preeclampsia in the Reduced Uterine Perfusion Pressure (RUPP) rat model. Free Radic Biol Med 2016, 101: 524-533.
- Masuda A, Ohno K. Neurodegeneration-associated RNA-binding protein, FUS, regulates mRNA length. Atlas of Science. Ed. by Lynn C Yeoman. AoS Nordic AB, Stockholm, 2016, http://atlasofscience.org/neurodegeneration-associated-rna-binding-protein-fus-regulates-mrna-length/ (査読有)
- Ohno K, Yagi H, Ohkawara B. Repositioning again of zonisamide for nerve regeneration. Neural Regener Res 2016, 11: 541-542. (査読有)
- Ohno K. Is the serum creatine kinase level elevated in congenital myasthenic syndrome? J Neurol Neurosurg Psychiatry 2016, 87: 801. (査読有)
- Ohno K, Ohkawara B, Ito M. Recent advances in congenital myasthenic syndromes. Clin Exp Neuroimmunol 2016, 7: 246–259. (査読有)
- Masuda A, Takeda J, Ohno K. FUS-mediated regulation of alternative RNA processing in neurons: insights from global transcriptome analysis. Wiley Interdiscip Rev RNA 2016, 7: 330-340. (査読有)
- Ohno K, Otsuka K, Ito M. Roles of collagen Q in MuSK antibody-positive myasthenia gravis. Chem Biol Interact 2016, 259: 266-270. (査読有)
- 2015年
- Azuma Y, Nakata T, Tanaka M, Shen XM, Ito M, Iwata S, Okuno T, Nomura Y, Ando N, Ishigaki K, Ohkawara B, Masuda A, Natsume J, Kojima S, Sokabe M, Ohno K. Congenital myasthenic syndrome in Japan: ethnically unique mutations in muscle nicotinic acetylcholine receptor subunits. Neuromuscul Disord, 2015; 25: 60-69.
- Matsushita M, Hasegawa S, Kitoh H, Mori K, Ohkawara B, Yasoda A, Masuda A, Ishiguro N, Ohno K. Meclozine promotes longitudinal skeletal growth in transgenic mice with achondroplasia carrying a gain-of-function mutation in the FGFR3 gene. Endocrinology, 2015; 156: 548-554.
- Tsunoda M, Hirayama M, Tsuda T, Ohno K. Noninvasive monitoring of plasma L-dopa concentrations using sweat samples in Parkinson's disease. Clin Chim Acta, 2015; 442: 52-55.
- Sobue S, Yamai K, Ito M, Ohno K, Ito M, Iwamoto T, Qiao S, Ohkuwa T, Ichihara M. Simultaneous oral and inhalational intake of molecular hydrogen additively suppresses signaling pathways in rodents. Mol Cell Biochem, 2015; 403: 231-241.
- Funayama M, Ohe K, Amo T, Furuya N, Yamaguchi J, Saiki S, Li Y, Ogaki K, Ando M, Yoshino H, Tomiyama H, Nishioka K, Hasegawa K, Saiki H, Satake W, Mogushi K, Sasaki R, Kokubo Y, Kuzuhara S, Toda T, Mizuno Y, Uchiyama Y, Ohno K, Hattori N. CHCHD2 mutations in autosomal dominant late-onset Parkinson's disease: a genome-wide linkage and sequencing study. Lancet Neurol, 2015; 14: 274-282.
- Selcen D, Ohkawara B, Shen XM, McEvoy K, Ohno K, Engel AG. Impaired Synaptic Development, Maintenance, and Neuromuscular Transmission in LRP4-Related Myasthenia. JAMA Neurol, 2015; 72: 889-896.
- Udagawa T, Fujioka Y, Tanaka M, Honda D, Yokoi S, Riku Y, Ibi D, Nagai T, Yamada K, Watanabe H, Katsuno M, Inada T, Ohno K, Sokabe M, Okado H, Ishigaki S, Sobue G. FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat Commun, 2015; 6: 7098.
- Fujii H, Matsubara K, Sakai K, Ito M, Ohno K, Ueda M, Yamamoto A. Dopaminergic differentiation of stem cells from human deciduous teeth and their therapeutic benefits for Parkinsonian rats. Brain Res, 2015; 1613: 59-72.
- Iwata S, Ito M, Nakata T, Noguchi Y, Okuno T, Ohkawara B, Masuda A, Goto T, Adachi M, Osaka H, Nonaka R, Arikawa-Hirasawa E, Ohno K. A missense mutation in domain III in HSPG2 in Schwartz-Jampel syndrome compromises secretion of perlecan into the extracellular space. Neuromuscul Disord, 2015; 25: 667-671.
- Kishimoto Y, Kato T, Ito M, Azuma Y, Fukasawa Y, Ohno K, Kojima S. Hydrogen ameliorates pulmonary hypertension in rats by anti-inflammatory and antioxidant effects. J Thorac Cardiovasc Surg, 2015; 150: 645-654 e643.
- Rahman MA, Azuma Y, Nasrin F, Takeda J, Nazim M, Bin Ahsan K, Masuda A, Engel AG, Ohno K. SRSF1 and hnRNP H antagonistically regulate splicing of COLQ exon 16 in a congenital myasthenic syndrome. Sci Rep, 2015; 5: 13208.
- Masuda A, Takeda J, Okuno T, Okamoto T, Ohkawara B, Ito M, Ishigaki S, Sobue G, Ohno K. Position-specific binding of FUS to nascent RNA regulates mRNA length. Genes Dev, 2015; 29: 1045-1057.
- Otsuka K, Ito M, Ohkawara B, Masuda A, Kawakami Y, Sahashi K, Nishida H, Mabuchi N, Takano A, Engel AG, Ohno K. Collagen Q and anti-MuSK autoantibody competitively suppress agrin/LRP4/MuSK signaling. Sci Rep, 2015; 5: 13928.
- Hasegawa S, Goto S, Tsuji H, Okuno T, Asahara T, Nomoto K, Shibata A, Fujisawa Y, Minato T, Okamoto A, Ohno K, Hirayama M. Intestinal Dysbiosis and Lowered Serum Lipopolysaccharide-Binding Protein in Parkinson's Disease. PLoS One, 2015; 10: e0142164.
- Yagi H, Ohkawara B, Nakashima H, Ito K, Tsushima M, Ishii H, Noto K, Ohta K, Masuda A, Imagama S, Ishiguro N, Ohno K. Zonisamide Enhances Neurite Elongation of Primary Motor Neurons and Facilitates Peripheral Nerve Regeneration In Vitro and in a Mouse Model. PLoS One, 2015; 10: e0142786.
- Mishima K, Kitoh H, Ohkawara B, Okuno T, Ito M, Masuda A, Ishiguro N, Ohno K. Lansoprazole upregulates polyubiquitination of TNF receptor associated factor 6 and facilitates Runx2-mediated osteoblastogenesis. EBioMedicine, 2015; 2: 2046-2061.
- Ito M, Ohno K. A hereditary mutation in Schwartz-Jampel syndrome. In: Atlas of Science, edited by LC Yeoman, 2015, AoS Nordic AB, Stockholm.
- Rahman MA, Ohno K. Splicing aberrations in congenital myasthenic syndromes. J Investig Genomics, 2015; 2: 00038.
- Rahman MA, Nasrin F, Masuda A, Ohno K. Decoding abnormal splicing code in human diseases. J Investig Genomics 2015; 2: 00016
- Ichihara M, Sobue S, Ito M, Ito M, Hirayama M, Ohno K. Beneficial biological effects and the underlying mechanisms of molecular hydrogen - comprehensive review of 321 original articles. Med Gas Res, 2015; 5: 12.
- 2014年
- Ohkawara B, Cabrera-Serrano M, Nakata T, Milone M, Asai N, Ito K, Ito M, Masuda A, Ito Y, Engel AG, Ohno K. LRP4 third beta-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum Mol Genet, 2014; 23: 1856-1868.
- Inaguma Y, Hamada N, Tabata H, Iwamoto I, Mizuno M, Nishimura YV, Ito H, Morishita R, Suzuki M, Ohno K, Kumagai T, Nagata K. SIL1, a causative cochaperone gene of Marinesco-Sojgren syndrome, plays an essential role in establishing the architecture of the developing cerebral cortex. EMBO Mol Med, 2014; 6: 414-429.
- Nakayama T, Nakamura H, Oya Y, Kimura T, Imahuku I, Ohno K, Nishino I, Abe K, Matsuura T. Clinical and genetic analysis of the first known Asian family with myotonic dystrophy type 2. J Hum Genet, 2014; 59: 129-133.
- Kokunai Y, Nakata T, Furuta M, Sakata S, Kimura H, Aiba T, Yoshinaga M, Osaki Y, Nakamori M, Itoh H, Sato T, Kubota T, Kadota K, Shindo K, Mochizuki H, Shimizu W, Horie M, Okamura Y, Ohno K, Takahashi MP. A Kir3.4 mutation causes Andersen-Tawil syndrome by an inhibitory effect on Kir2.1. Neurology, 2014; 82: 1058-1064.
- Mano Y, Kotani T, Ito M, Nagai T, Ichinohashi Y, Yamada K, Ohno K, Kikkawa F, Toyokuni S. Maternal molecular hydrogen administration ameliorates rat fetal hippocampal damage caused by in utero ischemia-reperfusion. Free Radic Biol Med, 2014; 69: 324-330.
- Takamatsu A, Ohkawara B, Ito M, Masuda A, Sakai T, Ishiguro N, Ohno K. Verapamil protects against cartilage degradation in osteoarthritis by inhibiting Wnt/beta-catenin signaling. PLoS One, 2014; 9: e92699.
- Kobayashi M, Ohno T, Ihara K, Murai A, Kumazawa M, Hoshino H, Iwanaga K, Iwai H, Hamana Y, Ito M, Ohno K, Horio F. Searching for genomic region of high-fat diet-induced type 2 diabetes in mouse chromosome 2 by analysis of congenic strains. PLoS One, 2014; 9: e96271.
- Yamashita Y, Matsuura T, Kurosaki T, Amakusa Y, Kinoshita M, Ibi T, Sahashi K, Ohno K. LDB3 splicing abnormalities are specific to skeletal muscles of patients with myotonic dystrophy type 1 and alter its PKC binding affinity. Neurobiol Dis, 2014; 69: 200-205.
- Asai N, Ohkawara B, Ito M, Masuda A, Ishiguro N, Ohno K. LRP4 induces extracellular matrix productions and facilitates chondrocyte differentiation. Biochem Biophys Res Commun, 2014; 451: 302-307.
- Nasrin F, Rahman MA, Masuda A, Ohe K, Takeda J, Ohno K. HnRNP C, YB-1 and hnRNP L coordinately enhance skipping of human MUSK exon 10 to generate a Wnt-insensitive MuSK isoform. Sci Rep, 2014; 4: 6841.
- Ohno K, Ohkawara B, Ito M, Engel AG. Molecular Genetics of Congenital Myasthenic Syndromes. In: eLS, 2014, John Wiley & Sons, Inc.
- Ohno K, Ito M, Kawakami Y, Ohtsuka K. Collagen Q is a key player for developing rational therapy for congenital myasthenia and for dissecting the mechanisms of anti-MuSK myasthenia gravis. J Mol Neurosci, 2014; 53: 359-361.
- Ohno K. Mutation analysis of a large cohort of GNE myopathy reveals a diverse array of GNE mutations affecting sialic acid biosynthesis. J Neurol Neurosurg Psychiatry, 2014; 85: 831.
- Noda M, Ito M, Ohsawa I, Ohno K. Beneficial effects of hydrogen in the CNS and a new brain-stomach interaction. Eur J Neurodeg Dis, 2014; 3: 25-34.
- Noda M, Fujita K, Ohsawa I, Ito M, Ohno K. Multiple effects of molecular hydrogen and its distinct mechanism. J Neurol Disord, 2014; 2: 6.
- 2013年
- Yamamoto R, Matsushita M, Kitoh H, Masuda A, Ito M, Katagiri T, Kawai T, Ishiguro N, Ohno K. Clinically applicable antianginal agents suppress osteoblastic transformation of myogenic cells and heterotopic ossifications in mice. J Bone Miner Metab, 2013; 31: 26-33.
- Sayeed S, Asano E, Ito S, Ohno K, Hamaguchi M, Senga T. S100A10 is required for the organization of actin stress fibers and promotion of cell spreading. Mol Cell Biochem, 2013; 374: 105-111.
- Iio A, Ito M, Itoh T, Terazawa R, Fujita Y, Nozawa Y, Ohsawa I, Ohno K, Ito M. Molecular hydrogen attenuates fatty acid uptake and lipid accumulation through downregulating CD36 expression in HepG2 cells. Med Gas Res, 2013; 3: 6.
- Nakata T, Ito M, Azuma Y, Otsuka K, Noguchi Y, Komaki H, Okumura A, Shiraishi K, Masuda A, Natsume J, Kojima S, Ohno K. Mutations in the C-terminal domain of ColQ in endplate acetylcholinesterase deficiency compromise ColQ-MuSK interaction. Hum Mutat, 2013; 34: 997-1004.
- Tanisawa K, Mikami E, Fuku N, Honda Y, Honda S, Ohsawa I, Ito M, Endo S, Ihara K, Ohno K, Kishimoto Y, Ishigami A, Maruyama N, Sawabe M, Iseki H, Okazaki Y, Hasegawa-Ishii S, Takei S, Shimada A, Hosokawa M, Mori M, Higuchi K, Takeda T, Higuchi M, Tanaka M. Exome sequencing of senescence-accelerated mice (SAM) reveals deleterious mutations in degenerative disease-causing genes. BMC Genomics, 2013; 14: 248.
- Selcen D, Shen XM, Milone M, Brengman J, Ohno K, Deymeer F, Finkel R, Rowin J, Engel AG. GFPT1-myasthenia: clinical, structural, and electrophysiologic heterogeneity. Neurology, 2013; 81: 370-378.
- Tsunoda M, Hirayama M, Ohno K, Tsuda T. A simple analytical method involving the use of a monolithic silica disk-packed spin column and HPLC-ECD for determination of L-DOPA in plasma of patients with Parkinson's disease. Analytical Methods, 2013; 5: 5161-5164.
- Fujioka Y, Ishigaki S, Masuda A, Iguchi Y, Udagawa T, Watanabe H, Katsuno M, Ohno K, Sobue G. FUS-regulated region- and cell-type-specific transcriptome is associated with cell selectivity in ALS/FTLD. Sci Rep, 2013; 3: 2388.
- Kitoh H, Achiwa M, Kaneko H, Mishima K, Matsushita M, Kadono I, Horowitz JD, Sallustio BC, Ohno K, Ishiguro N. Perhexiline maleate in the treatment of fibrodysplasia ossificans progressiva: an open-labeled clinical trial. Orphanet J Rare Dis, 2013; 8: 163.
- Matsushita M, Kitoh H, Ohkawara B, Mishima K, Kaneko H, Ito M, Masuda A, Ishiguro N, Ohno K. Meclozine facilitates proliferation and differentiation of chondrocytes by attenuating abnormally activated FGFR3 signaling in achondroplasia. PLoS One, 2013; 8: e81569.
- Rahman MA, Masuda A, Ohe K, Ito M, Hutchinson DO, Mayeda A, Engel AG, Ohno K. HnRNP L and hnRNP LL antagonistically modulate PTB-mediated splicing suppression of CHRNA1 pre-mRNA. Sci Rep, 2013; 3: 2931.
- Honda D, Ishigaki S, Iguchi Y, Fujioka Y, Udagawa T, Masuda A, Ohno K, Katsuno M, Sobue G. The ALS/FTLD-related RNA-binding proteins TDP-43 and FUS have common downstream RNA targets in cortical neurons. FEBS Open Bio, 2013; 4: 1-10.
- Ohe K, Masuda A, Ohno K. Intronic and exonic nucleotide variations that affect RNA splicing in humans. In: Genomics I – Humans, Animals and Plants. pp. 29-46, 2013, iConcept Press.
- Ohno K, Ito M, Kawakami Y, Krejci E, Engel AG. Specific binding of collagen Q to the neuromuscular junction is exploited to cure congenital myasthenia and to explore bases of myasthenia gravis. Chem Biol Interact, 2013; 203: 335-340.
- Ohno K. Glycosylation defects as an emerging novel cause leading to a limb-girdle type of congenital myasthenic syndromes. J Neurol Neurosurg Psychiatry, 2013; 84: 1064.
- 2012年
- Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, Ohno K. CUGBP1 and MBNL1 preferentially bind to 3' UTRs and facilitate mRNA decay. Sci Rep, 2012; 2: 209.
- Yoshinaga H, Sakoda S, Good JM, Takahashi MP, Kubota T, Arikawa-Hirasawa E, Nakata T, Ohno K, Kitamura T, Kobayashi K, Ohtsuka Y. A novel mutation in SCN4A causes severe myotonia and school-age-onset paralytic episodes. J Neurol Sci, 2012; 315: 15-19.
- Ito M, Suzuki Y, Okada T, Fukudome T, Yoshimura T, Masuda A, Takeda S, Krejci E, Ohno K. Protein-anchoring strategy for delivering acetylcholinesterase to the neuromuscular junction. Mol Ther, 2012; 20: 1384-1392.
- Ito M, Hirayama M, Yamai K, Goto S, Ito M, Ichihara M, Ohno K. Drinking hydrogen water and intermittent hydrogen gas exposure, but not lactulose or continuous hydrogen gas exposure, prevent 6-hydorxydopamine-induced Parkinson's disease in rats. Med Gas Res, 2012; 2: 15.
- Kurosaki T, Ueda S, Ishida T, Abe K, Ohno K, Matsuura T. The unstable CCTG repeat responsible for myotonic dystrophy type 2 originates from an AluSx element insertion into an early primate genome. PLoS One, 2012; 7: e38379.
- Ishigaki S, Masuda A, Fujioka Y, Iguchi Y, Katsuno M, Shibata A, Urano F, Sobue G, Ohno K. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci Rep, 2012; 2: 529.
- Yamashita Y, Matsuura T, Shinmi J, Amakusa Y, Masuda A, Ito M, Kinoshita M, Furuya H, Abe K, Ibi T, Sahashi K, Ohno K. Four parameters increase the sensitivity and specificity of the exon array analysis and disclose 25 novel aberrantly spliced exons in myotonic dystrophy. J Hum Genet, 2012; 57: 368-374.
- Matsuura T, Minami N, Arahata H, Ohno K, Abe K, Hayashi YK, Nishino I. Myotonic dystrophy type 2 is rare in the Japanese population. J Hum Genet, 2012; 57: 219-220.
- Engel AG, Shen X-M, Ohno K, Sine SM. Congenital myasthenic syndromes. In: Myasthenia gravis and myasthenic disorders 2nd ed., edited by AG Engel. pp. 173-230, 2012, Oxford University Press, New York.
- Ohno K, Ito M, Engel AG. Congenital Myasthenic Syndromes -Molecular Bases of Congenital Defects of Proteins at the Neuromuscular Junction- In: Neuromuscul Disord. pp. 175-200, 2012, InTech, Rijeka.
- Ohno K, Ito M, Ichihara M, Ito M. Molecular hydrogen as an emerging therapeutic medical gas for neurodegenerative and other diseases. Oxid Med Cell Longev, 2012; 2012: 353152.
- 2011年
- Selcen D, Juel VC, Hobson-Webb LD, Smith EC, Stickler DE, Bite AV, Ohno K, Engel AG. Myasthenic syndrome caused by plectinopathy. Neurology, 2011; 76: 327-336.
- Hirayama M, Nakamura T, Watanabe H, Uchida K, Hama T, Hara T, Niimi Y, Ito M, Ohno K, Sobue G. Urinary 8-hydroxydeoxyguanosine correlate with hallucinations rather than motor symptoms in Parkinson's disease. Parkinsonism Relat Disord, 2011; 17: 46-49.
- Fu Y, Masuda A, Ito M, Shinmi J, Ohno K. AG-dependent 3'-splice sites are predisposed to aberrant splicing due to a mutation at the first nucleotide of an exon. Nucleic Acids Res, 2011; 39: 4396-4404.
- Itoh T, Hamada N, Terazawa R, Ito M, Ohno K, Ichihara M, Nozawa Y, Ito M. Molecular hydrogen inhibits lipopolysaccharide/interferon gamma-induced nitric oxide production through modulation of signal transduction in macrophages. Biochem Biophys Res Commun, 2011; 411: 143-149.
- Kaneko H, Kitoh H, Matsuura T, Masuda A, Ito M, Mottes M, Rauch F, Ishiguro N, Ohno K. Hyperuricemia cosegregating with osteogenesis imperfecta is associated with a mutation in GPATCH8. Hum Genet, 2011; 130: 671-683.
- Ito M, Ibi T, Sahashi K, Ichihara M, Ito M, Ohno K. Open-label trial and randomized, double-blind, placebo-controlled, crossover trial of hydrogen-enriched water for mitochondrial and inflammatory myopathies. Med Gas Res, 2011; 1: 24.
- Kawakami Y, Ito M, Hirayama M, Sahashi K, Ohkawara B, Masuda A, Nishida H, Mabuchi N, Engel AG, Ohno K. Anti-MuSK autoantibodies block binding of collagen Q to MuSK. Neurology, 2011; 77: 1819-1826.
- Ohno K, Engel AG. Molecular defects of acetylcholine receptor subunits in congenital myasthenic syndromes. In: Pharmacology of Nicotinic Acetylcholine Receptors from the Basic and Therapeutic Perspectives, edited by HR Arias. pp. 175-186, 2011, Research Signpost, Kerala.
- Ohno K, Masuda A. RNA pathologies in neurological disorders. In: Neurochemical Mechanisms in Disease, edited by JP Blass. pp. 399-415, 2011, Springer, New York.
- Ohta S, Nakao A, Ohno K. The 2011 Medical Molecular Hydrogen Symposium: An inaugural symposium of the journal Medical Gas Research. Med Gas Res, 2011; 1: 10.
- 2009年
- Fu Y, Ito M, Fujita Y, Ito M, Ichihara M, Masuda A, Suzuki Y, Maesawa S, Kajita Y, Hirayama M, Ohsawa I, Ohta S, Ohno K. Molecular hydrogen is protective against 6-hydroxydopamine-induced nigrostriatal degeneration in a rat model of Parkinson's disease. Neurosci Lett, 2009; 453: 81-85.
- Milone M, Shen XM, Selcen D, Ohno K, Brengman J, Iannaccone ST, Harper CM, Engel AG. Myasthenic syndrome due to defects in rapsyn: Clinical and molecular findings in 39 patients. Neurology, 2009; 73: 228-235.
- Bian Y, Masuda A, Matsuura T, Ito M, Okushin K, Engel AG, Ohno K. Tannic acid facilitates expression of the polypyrimidine tract binding protein and alleviates deleterious inclusion of CHRNA1 exon P3A due to an hnRNP H-disrupting mutation in congenital myasthenic syndrome. Hum Mol Genet, 2009; 18: 1229-1237.
- Almeida T, Alonso I, Martins S, Ramos EM, Azevedo L, Ohno K, Amorim A, Saraiva-Pereira ML, Jardim LB, Matsuura T, Sequeiros J, Silveira I. Ancestral origin of the ATTCT repeat expansion in spinocerebellar ataxia type 10 (SCA10). PLoS One, 2009; 4: e4553.
- Kurosaki T, Matsuura T, Ohno K, Ueda S. Alu-mediated acquisition of unstable ATTCT pentanucleotide repeats in the human ATXN10 gene. Mol Biol Evol, 2009; 26: 2573-2579.
- Itoh T, Fujita Y, Ito M, Masuda A, Ohno K, Ichihara M, Kojima T, Nozawa Y, Ito M. Molecular hydrogen suppresses FcepsilonRI-mediated signal transduction and prevents degranulation of mast cells. Biochem Biophys Res Commun, 2009; 389: 651-656.
- 2008年
- Saito T, Amakusa Y, Kimura T, Yahara O, Aizawa H, Ikeda Y, Day JW, Ranum LP, Ohno K, Matsuura T. Myotonic dystrophy type 2 in Japan: ancestral origin distinct from Caucasian families. Neurogenetics, 2008; 9: 61-63.
- Gao K, Masuda A, Matsuura T, Ohno K. Human branch point consensus sequence is yUnAy. Nucleic Acids Res, 2008; 36: 2257-2267.
- Kurosaki T, Matsuura T, Ohno K, Ueda S. Long-range PCR for the diagnosis of spinocerebellar ataxia type 10. Neurogenetics, 2008; 9: 151-152.
- Shen XM, Fukuda T, Ohno K, Sine SM, Engel AG. Congenital myasthenia-related AChR delta subunit mutation interferes with intersubunit communication essential for channel gating. J Clin Invest, 2008; 118: 1867-1876.
- Ito M, Masuda A, Jinno S, Katagiri T, Krejci E, Ohno K. Viral vector-mediated [corrected] expression of human collagen Q in cultured cells. Chem Biol Interact, 2008; 175: 346-348.
- Masuda A, Shen XM, Ito M, Matsuura T, Engel AG, Ohno K. hnRNP H enhances skipping of a nonfunctional exon P3A in CHRNA1 and a mutation disrupting its binding causes congenital myasthenic syndrome. Hum Mol Genet, 2008; 17: 4022-4035.
- 2007年
- Masuda A, Hashimoto K, Yokoi T, Doi T, Kodama T, Kume H, Ohno K, Matsuguchi T. Essential role of GATA transcriptional factors in the activation of mast cells. J Immunol, 2007; 178: 360-368.
- Ichihara M, Murakumo Y, Masuda A, Matsuura T, Asai N, Jijiwa M, Ishida M, Shinmi J, Yatsuya H, Qiao S, Takahashi M, Ohno K. Thermodynamic instability of siRNA duplex is a prerequisite for dependable prediction of siRNA activities. Nucleic Acids Res, 2007; 35: e123.
- Sahashi K, Masuda A, Matsuura T, Shinmi J, Zhang Z, Takeshima Y, Matsuo M, Sobue G, Ohno K. In vitro and in silico analysis reveals an efficient algorithm to predict the splicing consequences of mutations at the 5' splice sites. Nucleic Acids Res, 2007; 35: 5995-6003.
- 2005年
- Shen XM, Ohno K, Sine SM, Engel AG. Subunit-specific contribution to agonist binding and channel gating revealed by inherited mutation in muscle acetylcholine receptor M3-M4 linker. Brain, 2005; 128: 345-355.
- Ohno K, Tsujino A, Shen XM, Milone M, Engel AG. Spectrum of splicing errors caused by CHRNE mutations affecting introns and intron/exon boundaries. J Med Genet, 2005; 42: e53.
- Ohno K, Engel AG. Splicing abnormalities in congenital myasthenic syndromes. Acta Myol, 2005; 24: 50-54.
- 2004年
- Selcen D, Ohno K, Engel AG. Myofibrillar myopathy: clinical, morphological and genetic studies in 63 patients. Brain, 2004; 127: 439-451.
- Sahashi K, Ibi T, Ohno K, Sahashi K, Nakao N, Kondo H. Progressive myopathy with circulating autoantibody against giantin in the Golgi apparatus. Neurology, 2004; 62: 1891-1893.
- Ohno K, Engel AG. Lack of founder haplotype for the rapsyn N88K mutation: N88K is an ancient founder mutation or arises from multiple founders. J Med Genet, 2004; 41: e8.
- Kimbell LM, Ohno K, Engel AG, Rotundo RL. C-terminal and heparin-binding domains of collagenic tail subunit are both essential for anchoring acetylcholinesterase at the synapse. J Biol Chem, 2004; 279: 10997-11005.
- Cai Y, Cronin CN, Engel AG, Ohno K, Hersh LB, Rodgers DW. Choline acetyltransferase structure reveals distribution of mutations that cause motor disorders. EMBO J, 2004; 23: 2047-2058.
- Banwell BL, Ohno K, Sieb JP, Engel AG. Novel truncating RAPSN mutations causing congenital myasthenic syndrome responsive to 3,4-diaminopyridine. Neuromuscul Disord, 2004; 14: 202-207.
- Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes (Chapter 66). In: Myology, edited by AG Engel, C Franzini-Armstrong. pp. 1801-1844, 2004, McGraw Hill, New York.
- Sine SM, Engel AG, Wang H-L, Ohno K. Molecular Insights into Acetylcholine Receptor Structure and Function Revealed by Mutations Causing Congenital Myasthenic Syndromes. In: Molecular and Cellular Insights into Ion Channel Biology, edited by RA Maue. pp. 95-119, 2004, Elsevier Science, Amsterdam.
- 2003年
- Ohno K, Milone M, Shen XM, Engel AG. A frameshifting mutation in CHRNE unmasks skipping of the preceding exon. Hum Mol Genet, 2003; 12: 3055-3066.
- Ohno K, Sadeh M, Blatt I, Brengman JM, Engel AG. E-box mutations in the RAPSN promoter region in eight cases with congenital myasthenic syndrome. Hum Mol Genet, 2003; 12: 739-748.
- Tsujino A, Maertens C, Ohno K, Shen XM, Fukuda T, Harper CM, Cannon SC, Engel AG. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci U S A, 2003; 100: 7377-7382.
- Shen XM, Ohno K, Tsujino A, Brengman JM, Gingold M, Sine SM, Engel AG. Mutation causing severe myasthenia reveals functional asymmetry of AChR signature cystine loops in agonist binding and gating. J Clin Invest, 2003; 111: 497-505.
- Engel AG, Ohno K, Harper CM. Congenital myasthenic syndromes. In: Neuromuscular Disorders of Infancy, Childhood, and Adolescence: A Clinician's Approach, edited by HR Jones, C De Vivo D, BT Darras. pp. 555-574, 2003, Butterworth and Heinemann, Boston.
- Sine SM, Wang HL, Ohno K, Shen XM, Lee WY, Engel AG. Mechanistic diversity underlying fast channel congenital myasthenic syndromes. Ann N Y Acad Sci, 2003; 998: 128-137.
- Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes: A diverse array of molecular targets. J Neurocytol, 2003; 32: 1017-1037.
- Engel AG, Ohno K, Sine SM. Sleuthing molecular targets for neurological diseases at the neuromuscular junction. Nat Rev Neurosci, 2003; 4: 339-352.
- Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes: progress over the past decade. Muscle Nerve, 2003; 27: 4-25.
- Engel AG, Ohno K, Shen XM, Sine SM. Congenital myasthenic syndromes: Multiple molecular targets at the neuromuscular junction. Myasthenia Gravis and Related Disorders, 2003; 998: 138-160.
- 2002年
- Shen XM, Ohno K, Fukudome T, Tsujino A, Brengman JM, De Vivo DC, Packer RJ, Engel AG. Congenital myasthenic syndrome caused by low-expressor fast-channel AChR delta subunit mutation. Neurology, 2002; 59: 1881-1888.
- Shapira YA, Sadeh ME, Bergtraum MP, Tsujino A, Ohno K, Shen XM, Brengman J, Edwardson S, Matoth I, Engel AG. Three novel COLQ mutations and variation of phenotypic expressivity due to G240X. Neurology, 2002; 58: 603-609.
- Ohno K, Engel AG, Shen XM, Selcen D, Brengman J, Harper CM, Tsujino A, Milone M. Rapsyn mutations in humans cause endplate acetylcholine-receptor deficiency and myasthenic syndrome. Am J Hum Genet, 2002; 70: 875-885.
- Sine SM, Shen XM, Wang HL, Ohno K, Lee WY, Tsujino A, Brengmann J, Bren N, Vajsar J, Engel AG. Naturally occurring mutations at the acetylcholine receptor binding site independently alter ACh binding and channel gating. J Gen Physiol, 2002; 120: 483-496.
- Byring RF, Pihko H, Tsujino A, Shen XM, Gustafsson B, Hackman P, Ohno K, Engel AG, Udd B. Congenital myasthenic syndrome associated with episodic apnea and sudden infant death. Neuromuscul Disord, 2002; 12: 548-553.
- Engel AG, Ohno K. Congenital myasthenic syndromes (Chapter 13). In: Adv Neurol, edited by R Pourmand, Y Harati. pp. 203-215, 2002, Lippincott Williams & Wilkins, Philadelphia.
- Engel AG, Ohno K, Selcen D. Congenital Myasthenic Syndromes. In: Structural and Molecular Basis of Skeletal Muscle Diseases, edited by G Karpati. pp. 170-179, 2002, International Society of Neuropathology/World Federation of Neurology. ISN Neuropath Press, Basel.
- Ohno K, Engel AG. Congenital myasthenic syndromes: genetic defects of the neuromuscular junction. Curr Neurol Neurosci Rep, 2002; 2: 78-88.
- Engel AG, Ohno K, Sine SM. The spectrum of congenital myasthenic syndromes. Mol Neurobiol, 2002; 26: 347-367.
- 2001年
- Sahashi K, Yoneda M, Ohno K, Tanaka M, Ibi T, Sahashi K. Functional characterisation of mitochondrial tRNA(Tyr) mutation (5877-->GA) associated with familial chronic progressive external ophthalmoplegia. J Med Genet, 2001; 38: 703-705.
- Ohno K, Tsujino A, Brengman JM, Harper CM, Bajzer Z, Udd B, Beyring R, Robb S, Kirkham FJ, Engel AG. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci U S A, 2001; 98: 2017-2022.
- Engel AG, Ohno K, Sine SM. Acetylcholine receptor channelopathies and other congenital myasthenic syndromes (Chapter 12). In: Channelopathies of the nervous system, edited by MR Rose, RC Griggs. pp. 179-191, 2001, Butterworth and Heinemann, Boston.
- 2000年
- Ohno K, Engel AG, Brengman JM, Shen XM, Heidenreich F, Vincent A, Milone M, Tan E, Demirci M, Walsh P, Nakano S, Akiguchi I. The spectrum of mutations causing end-plate acetylcholinesterase deficiency. Ann Neurol, 2000; 47: 162-170.
- Wang HL, Ohno K, Milone M, Brengman JM, Evoli A, Batocchi AP, Middleton LT, Christodoulou K, Engel AG, Sine SM. Fundamental gating mechanism of nicotinic receptor channel revealed by mutation causing a congenital myasthenic syndrome. J Gen Physiol, 2000; 116: 449-462.
- Engel AG, Ohno K, Stans AA. Congenital myasthenic syndromes. In: Neuromuscular Diseases: From Basic Mechanisms To Clinical Management, edited by F Demeer. pp. 96-112, 2000, Karger, Basel.
- Engel AG, Ohno K, Shen XM, Milone M, Tsujino A. Congenital myasthenic syndromes in the molecular era. Acta Myol, 2000; 19: 5-21.
- 1999年
- Middleton L, Ohno K, Christodoulou K, Brengman J, Milone M, Neocleous V, Serdaroglu P, Deymeer F, Ozdemir C, Mubaidin A, Horany K, Al-Shehab A, Mavromatis I, Mylonas I, Tsingis M, Zamba E, Pantzaris M, Kyriallis K, Engel AG. Chromosome 17p-linked myasthenias stem from defects in the acetylcholine receptor epsilon-subunit gene. Neurology, 1999; 53: 1076-1082.
- Ohno K, Brengman JM, Felice KJ, Cornblath DR, Engel AG. Congenital end-plate acetylcholinesterase deficiency caused by a nonsense mutation and an A-->G splice-donor-site mutation at position +3 of the collagenlike-tail-subunit gene (COLQ): how does G at position +3 result in aberrant splicing? Am J Hum Genet, 1999; 65: 635-644.
- Wang HL, Milone M, Ohno K, Shen XM, Tsujino A, Batocchi AP, Tonali P, Brengman J, Engel AG, Sine SM. Acetylcholine receptor M3 domain: stereochemical and volume contributions to channel gating. Nat Neurosci, 1999; 2: 226-233.
- Quiram PA, Ohno K, Milone M, Patterson MC, Pruitt NJ, Brengman JM, Sine SM, Engel AG. Mutation causing congenital myasthenia reveals acetylcholine receptor beta/delta subunit interaction essential for assembly. J Clin Invest, 1999; 104: 1403-1410.
- Ohno K, Anlar B, Engel AG. Congenital myasthenic syndrome caused by a mutation in the Ets-binding site of the promoter region of the acetylcholine receptor epsilon subunit gene. Neuromuscul Disord, 1999; 9: 131-135.
- Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes (Chapter 11). In: Myasthenia gravis and myasthenic disorders, edited by AG Engel. pp. 251-297, 1999, Oxford University Press, New York.
- Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes: recent advances. Arch Neurol, 1999; 56: 163-167.
- 1998年
- Ohno K, Anlar B, Ozdirim E, Brengman JM, DeBleecker JL, Engel AG. Myasthenic syndromes in Turkish kinships due to mutations in the acetylcholine receptor. Ann Neurol, 1998; 44: 234-241.
- Ohno K, Anlar B, Ozdirim E, Brengman JM, Engel AG. Frameshifting and splice-site mutations in the acetylcholine receptor epsilon subunit gene in three Turkish kinships with congenital myasthenic syndromes. Ann N Y Acad Sci, 1998; 841: 189-194.
- Milone M, Ohno K, Fukudome T, Shen XM, Brengman J, Griggs RC, Engel AG. Congenital myasthenic syndrome caused by novel loss-of-function mutations in the human AChR epsilon subunit gene. Ann N Y Acad Sci, 1998; 841: 184-188.
- Fukudome T, Ohno K, Brengman JM, Engel AG. AChR channel blockade by quinidine sulfate reduces channel open duration in the slow-channel congenital myasthenic syndrome. Ann N Y Acad Sci, 1998; 841: 199-202.
- Ohno K, Brengman J, Tsujino A, Engel AG. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proc Natl Acad Sci U S A, 1998; 95: 9654-9659.
- Milone M, Wang HL, Ohno K, Prince R, Fukudome T, Shen XM, Brengman JM, Griggs RC, Sine SM, Engel AG. Mode switching kinetics produced by a naturally occurring mutation in the cytoplasmic loop of the human acetylcholine receptor epsilon subunit. Neuron, 1998; 20: 575-588.
- Fukudome T, Ohno K, Brengman JM, Engel AG. Quinidine normalizes the open duration of slow-channel mutants of the acetylcholine receptor. Neuroreport, 1998; 9: 1907-1911.
- Ohno K, Engel AG. Congenital myasthenic syndromes: gene mutation. Neuromuscular disorders : NMD, 1998; 8: XII-XIII.
- Engel AG, Ohno K, Wang HL, Milone M, Sine SM. Molecular basis of congenital myasthenic syndromes: Mutations in the acetylcholine receptor. Neuroscientist, 1998; 4: 185-194.
- Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes: experiments of nature. J Physiol Paris, 1998; 92: 113-117.
- Engel AG, Ohno K, Milone M, Sine SM. Congenital myasthenic syndromes. New insights from molecular genetic and patch-clamp studies. Ann N Y Acad Sci, 1998; 841: 140-156.
- 1997年
- Wang HL, Auerbach A, Bren N, Ohno K, Engel AG, Sine SM. Mutation in the M1 domain of the acetylcholine receptor alpha subunit decreases the rate of agonist dissociation. J Gen Physiol, 1997; 109: 757-766.
- Milone M, Wang HL, Ohno K, Fukudome T, Pruitt JN, Bren N, Sine SM, Engel AG. Slow-channel myasthenic syndrome caused by enhanced activation, desensitization, and agonist binding affinity attributable to mutation in the M2 domain of the acetylcholine receptor alpha subunit. J Neurosci, 1997; 17: 5651-5665.
- Ohno K, Quiram PA, Milone M, Wang HL, Harper MC, Pruitt JN, 2nd, Brengman JM, Pao L, Fischbeck KH, Crawford TO, Sine SM, Engel AG. Congenital myasthenic syndromes due to heteroallelic nonsense/missense mutations in the acetylcholine receptor epsilon subunit gene: identification and functional characterization of six new mutations. Hum Mol Genet, 1997; 6: 753-766.
- 1996年
- Engel AG, Ohno K, Milone M, Sine SM. Congenital myasthenic syndromes caused by mutations in acetylcholine receptor genes. Neurology, 1997; 48: 28S-35S.
- Sawano T, Tanaka M, Ohno K, Yoneda M, Ota Y, Terasaki H, Awaya S, Ozawa T. Mitochondrial DNA mutations associated with the 11778 mutation in Leber's disease. Biochem Mol Biol Int, 1996; 38: 693-700.
- Ohno K, Yamamoto M, Engel AG, Harper CM, Roberts LR, Tan GH, Fatourechi V. MELAS- and Kearns-Sayre-type co-mutation [corrected] with myopathy and autoimmune polyendocrinopathy. Ann Neurol, 1996; 39: 761-766.
- Engel AG, Ohno K, Bouzat C, Sine SM, Griggs RC. End-plate acetylcholine receptor deficiency due to nonsense mutations in the epsilon subunit. Ann Neurol, 1996; 40: 810-817.
- Ohno K, Wang HL, Milone M, Bren N, Brengman JM, Nakano S, Quiram P, Pruitt JN, Sine SM, Engel AG. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit. Neuron, 1996; 17: 157-170.
- Engel AG, Ohno K, Milone M, Wang HL, Nakano S, Bouzat C, Pruitt JN, 2nd, Hutchinson DO, Brengman JM, Bren N, Sieb JP, Sine SM. New mutations in acetylcholine receptor subunit genes reveal heterogeneity in the slow-channel congenital myasthenic syndrome. Hum Mol Genet, 1996; 5: 1217-1227.
- 1995年
- Ohno K, Hutchinson DO, Milone M, Brengman JM, Bouzat C, Sine SM, Engel AG. Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the epsilon subunit. Proc Natl Acad Sci U S A, 1995; 92: 758-762.
- Sine SM, Ohno K, Bouzat C, Auerbach A, Milone M, Pruitt JN, Engel AG. Mutation of the acetylcholine receptor alpha subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron, 1995; 15: 229-239.
- 1993年
- Sano T, Ban K, Ichiki T, Kobayashi M, Tanaka M, Ohno K, Ozawa T. Molecular and genetic analyses of two patients with Pearson's marrow-pancreas syndrome. Pediatr Res, 1993; 34: 105-110.
- Suoh H, Sahashi K, Ibi T, Tashiro M, Tanaka F, Mitsuma T, Ohno K. Progressive external ophthalmoplegia and myositis. Intern Med, 1993; 32: 319-322.
- 1992年
- Sahashi K, Tanaka M, Tashiro M, Ohno K, Ibi T, Takahashi A, Ozawa T. Increased mitochondrial DNA deletions in the skeletal muscle of myotonic dystrophy. Gerontology, 1992; 38: 18-29.
- 1991年
- Ohno K, Tanaka M, Sahashi K, Ibi T, Sato W, Yamamoto T, Takahashi A, Ozawa T. Mitochondrial DNA deletions in inherited recurrent myoglobinuria. Ann Neurol, 1991; 29: 364-369.
- Tanaka M, Ino H, Ohno K, Ohbayashi T, Ikebe S, Sano T, Ichiki T, Kobayashi M, Wada Y, Ozawa T. Mitochondrial DNA mutations in mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Biochem Biophys Res Commun, 1991; 174: 861-868.
- Ozawa T, Tanaka M, Ino H, Ohno K, Sano T, Wada Y, Yoneda M, Tanno Y, Miyatake T, Tanaka T, et al. Distinct clustering of point mutations in mitochondrial DNA among patients with mitochondrial encephalomyopathies and with Parkinson's disease. Biochem Biophys Res Commun, 1991; 176: 938-946.
- Ozawa T, Tanaka M, Sugiyama S, Ino H, Ohno K, Hattori K, Ohbayashi T, Ito T, Deguchi H, Kawamura K, et al. Patients with idiopathic cardiomyopathy belong to the same mitochondrial DNA gene family of Parkinson's disease and mitochondrial encephalomyopathy. Biochem Biophys Res Commun, 1991; 177: 518-525.
- Ohno K, Tanaka M, Suzuki H, Ohbayashi T, Ikebe S, Ino H, Kumar S, Takahashi A, Ozawa T. Identification of a possible control element, Mt5, in the major noncoding region of mitochondrial DNA by intraspecific nucleotide conservation. Biochem Int, 1991; 24: 263-272.
- Ota Y, Tanaka M, Sato W, Ohno K, Yamamoto T, Maehara M, Negoro T, Watanabe K, Awaya S, Ozawa T. Detection of platelet mitochondrial DNA deletions in Kearns-Sayre syndrome. Invest Ophthalmol Vis Sci, 1991; 32: 2667-2675.
- Ino H, Tanaka M, Ohno K, Hattori K, Ikebe S, Sano T, Ozawa T, Ichiki T, Kobayashi M, Wada Y. Mitochondrial leucine tRNA mutation in a mitochondrial encephalomyopathy. Lancet, 1991; 337: 234-235.
- Ohno K, Tanaka M, Ino H, Suzuki H, Tashiro M, Ibi T, Sahashi K, Takahashi A, Ozawa T. Direct DNA sequencing from colony: analysis of multiple deletions of mitochondrial genome. Biochim Biophys Acta, 1991; 1090: 9-16.
- Ozawa T, Tanaka M, Hayakawa M, Sugiyama S, Ino H, Sato W, Ohno K, Ikebe S, Yoneda M. Mitochondrial DNA disease: phylogeny and expression. In: New Era of Bioenergetics, edited by Y Mukohata. pp. 247-272, 1991, Academic Press, Tokyo.
- Ozawa T, Tanaka M, Hayakawa M, Sugiyama S, Sato W, Ohno K, Ikebe S, Yoneda M. Mitochondrial DNA mutations: types, mechanism and expression. In: Progress in Neuropathology Vol. 7, Mitochondrial Encephalomyopathies, edited by T Sato. pp. 141-151, 1991, Raven Press, New York.
- Ozawa T, Tanaka M, Sato W, Ohno K, Yoneda M. Diseases caused by mitochondrial DNA mutations: types and mechanism. In: Proceedings of the XIth International Congress of Neuropathology. pp. 481-485, 1991, Jpn. Soc. Neuropathol., Tokyo.
- Ozawa T, Tanaka M, Sato W, Ohno K, Yoneda M, Yamamoto T. Types and mechanism of mitochondrial DNA mutations in mitochondrial myopathy and related diseases. In: Molecular Basis of Neurological Disorders and their Treatment, edited by JW Gorrod, O Albano, E Ferrari, S Papa. pp. 173-190, 1991, Chapman and Hall, London.
- Sahashi K, Ibi T, Ohno K, Tanaka M, Tashiro M, Tsuchiya I, Nakao M, Yuasa K, Mitsuma T, Takahashi A, Ozawa T. Visceral myopathy with external ophthalmoplegia and multiple mitochondrial DNA deletions. In: New Trends in Autonomic Nervous System Research, edited by Mea Yoshikawa. pp. 229-230, 1991, Elsevier Science Publishers, B. V.
- Tanaka M, Hattori K, Ito H, Ohbayashi T, Ohno K, Sato W, Sugiyama S, Ozawa T. Mitochondrial DNA mutations in idiopathic cardiomyopathy and in presbycardia. In: Mitochondrial Encephalomyopathies, edited by T Sato. pp. 225-236, 1991, Raven Press, New York.
- 1990年
- Ozawa T, Tanaka M, Sugiyama S, Hattori K, Ito T, Ohno K, Takahashi A, Sato W, Takada G, Mayumi B, et al. Multiple mitochondrial DNA deletions exist in cardiomyocytes of patients with hypertrophic or dilated cardiomyopathy. Biochem Biophys Res Commun, 1990; 170: 830-836.
- Ikebe S, Tanaka M, Ohno K, Sato W, Hattori K, Kondo T, Mizuno Y, Ozawa T. Increase of deleted mitochondrial DNA in the striatum in Parkinson's disease and senescence. Biochem Biophys Res Commun, 1990; 170: 1044-1048.
- Ozawa T, Tanaka M, Ikebe S, Ohno K, Kondo T, Mizuno Y. Quantitative determination of deleted mitochondrial DNA relative to normal DNA in parkinsonian striatum by a kinetic PCR analysis. Biochem Biophys Res Commun, 1990; 172: 483-489.
- Sahashi K, Ohno K, Tanaka M, Ibi T, Yamamoto T, Tashiro M, Sato W, Takahashi A, Ozawa T. Cytoplasmic body and mitochondrial DNA deletion. J Neurol Sci, 1990; 99: 291-300.
- Tanaka M, Ino H, Ohno K, Hattori K, Sato W, Ozawa T, Tanaka T, Itoyama S. Mitochondrial mutation in fatal infantile cardiomyopathy. Lancet, 1990; 336: 1452.
- Ozawa T, Tanaka M, Sato W, Ohno K, Sugiyama S, Yoneda M, Yamamoto T, Hattori K, Ikebe S, Tashiro M, Sahashi K. Mitochondrial DNA mutations as an etiology of human degenerative diseases. In: Bioenergetics: Molecular Biology, Biochemistry, and Pathology, edited by CH Kim, T Ozawa. pp. 413-427, 1990, Plenum, New York and London.
- Tanaka M, ., Sato W, Ohno K, Yamamoto T, Ozawa T. S1 nuclease analysis and direct sequencing of deleted mitochondrial DNA in myopathic patients: Role of directly repeated sequences in deletion. In: Bioenergetics: Molecular Biology, Biochemistry, and Pathology, edited by CH Kim, T Ozawa. pp. 441-449, 1990, Plenum, New York.
- 1989年
- Sato W, Tanaka M, Ohno K, Yamamoto T, Takada G, Ozawa T. Multiple populations of deleted mitochondrial DNA detected by a novel gene amplification method. Biochem Biophys Res Commun, 1989; 162: 664-672.
- Tanaka M, Sato W, Ohno K, Yamamoto T, Ozawa T. Direct sequencing of deleted mitochondrial DNA in myopathic patients. Biochem Biophys Res Commun, 1989; 164: 156-163.
- Tanaka-Yamamoto T, Tanaka M, Ohno K, Sato W, Horai S, Ozawa T. Specific amplification of deleted mitochondrial DNA from a myopathic patient and analysis of deleted region with S1 nuclease. Biochim Biophys Acta, 1989; 1009: 151-155.
- Tanaka M, Yoneda M, Ohno K, Sato W, Yamamoto M, Nonaka I, Horai S, Ozawa T. Differently deleted mitochondrial genomes in maternally inherited chronic progressive external ophthalmoplegia. J Inherit Metab Dis, 1989; 12: 359-362.
- 1988年
- Ozawa T, Yoneda M, Tanaka M, Ohno K, Sato W, Suzuki H, Nishikimi M, Yamamoto M, Nonaka I, Horai S. Maternal inheritance of deleted mitochondrial DNA in a family with mitochondrial myopathy. Biochem Biophys Res Commun, 1988; 154: 1240-1247.
研究キーワード
先天性筋無力症候群、新経筋接合部、スプライシング、RNA結合タンパク、ドラッグリポジショニング、パーキンソン病、腸内細菌叢、分子状水素
大学院生募集
当研究室は、現在大学院生の募集を休止しております。